Курсовая работа
Выполнила студентка 4 курса, 541 группы Ревтович Светлана
Самарский Государственный Университет
Самара 2001
Введение
1. Обзор литературы
1.1. Отличия ДНК-белковых от РНК-белковых взаимодействий
Двойная спираль ДНК – это регулярная структура, в которой наиболее существенные отличия между соседними участками заключены в последовательности пар оснований. В обоих желобках специфический сайт ДНК для узнавания белком может быть сформирован сетью водородных связей между основаниями нуклеотидов. Большой желобок спирали ДНК доступнее для контакта, что создаёт возможность для дискриминации последовательностей в нём [39]. Основанный на этом предположении механизм узнавания «прямым считыванием» был подтверждён для многих ДНК-связывающих белков. Также было отмечено потенциальное значение и «непрямого считывания». Некоторые последовательности ДНК могут иметь изменения в конформации сахаро-фосфатного остова или даже быть деформированы в необычные структуры. В таком случае специфичность связывания может определяться контактами белка с остовом ДНК [40]. Индуцированные белком нарушения структуры ДНК довольно обычны и могут варьировать по форме от небольших изгибов, как в сайте нуклеазы EcoRI, до больших изгибов и выведения оснований из стэкинга, как в комплексе с ТАТА-связывающим белком [24].
Механизм «прямого считывания» предполагает, что ДНК-белковыми контактами, определяющими специфичность связывания, являются водородные связи с основаниями и гидрофобные контакты с тиминовыми метилами, в то время как энергия неспецифического связывания обеспечивается ионными и водородными связями с сахаро-фосфатным остовом. В «непрямом считывании» могут принимать участие и другие виды взаимодействий, например, в комплексе с ТАТА-связывающим белком ДНК контактирует с гидрофобной белковой поверхностью и ароматические аминокислоты интеркалируются в спираль ДНК [24].
Молекулы РНК отличаются от ДНК несколькими особенностями, которые влияют на возможности белкового узнавания. Во-первых, в РНК участки Уотсон-Криковских спиралей часто прерываются выпячиваниями, внутренними петлями и шпильками. Исследования часто встречающихся «четырёхнуклеотидных» шпилек и консервативных внутренних и шпилечных петель рибосомной РНК, методами ЯМР и рентгеноструктурного анализа выявили специфические нерегулярные структуры, содержащие неканонические пары и выпячивания. Во-вторых, спирали РНК достаточно короткие, обычно меньше полного оборота, и имеют А-форму, отличную от В-формы ДНК. А-форма спирали имеет очень глубокий и узкий большой желобок, что создаёт стерические затруднения для взаимодействия с белками. Однако прилегающий к нерегулярным участкам большой желобок может быть доступен для связывания [41].
Реклама

Следствием этих отличий является то, что белки оказываются перед значительно большим разнообразием водородных связей и возможностей стэкинга нуклеотидов в случае РНК, чем это возможно в стандартной спирали ДНК. Дополнительной и весьма важной особенностью РНК является возможность «третичных» взаимодействий, которые могут соединять различные участки РНК и создавать сложные структуры. Классическими примерами являются соединение спиралей тРНК в «L-образную» молекулу, обнаруженная в интронах группы I «аденозиновая платформа», открывающая спираль РНК для стэкинга с другой спиралью РНК [12], и структура двух взаимодействующих петлями транскриптов RNA I и RNA II плазмиды ColE1, так называемый «kissing» комплекс [27]. Тот факт, что благодаря вторичным и третичным взаимодействиям может быть создана уникальная структура РНК, повышает вероятность специфического взаимодействия белков только с сахаро-фосфатным остовом. Эта ситуация может быть крайним случаем «непрямого считывания», когда основания нуклеотидов обеспечивают белковое узнавание только определением общей конформации РНК, а не прямыми контактами с белком.
Таким образом, искажение регулярной структуры нуклеиновой кислоты, по всей видимости, более важно для узнавания РНК, чем для ДНК. Спираль ДНК довольно стабильная структура, в то время как неспаренные нуклеотиды и петли дестабилизируют структуру РНК. Комбинация богатого выбора нерегулярных структур в РНК и возможности деформации её структуры позволяют предположить, что РНК-связывающие белки будут использовать более широкий выбор стратегий связывания, чем ДНК-связывающие белки, и что механизм узнавания «непрямым считыванием» будет среди них более широко распространён.
1.2. Ранние представления об РНК-белковых взаимодействиях
1.2.1. РНК-связывающие структурные мотивы в белках
Изучение свойств РНК-связывающих белков привело к обнаружению в этих белках ряда и консервативных мотивов. Их открытие позволило начать классификацию РНК-связывающих белков, исходя из их структурных особенностей, а также предсказывать РНК-связывающие свойства белков [8]. В настоящее время выделяют следующие основные мотивы:
Реклама

РНП (ribonucleoprotein) мотив - наиболее распространён, состоит из двух коротких последовательностей РНП1 (октамер) и РНП2, разделённых примерно 30 аминокислотами [17].
S1 мотив – в состав входит пять антипараллельных b-тяжей. Ряд консервативных ароматических и заряженных аминокислот, расположенных в петле между тяжами b1 и b2, в середине b2, в конце b3 и в повороте между b4 и b5 находятся поблизости друг от друга и, вероятно, формируют РНК-связывающий участок домена [10].
Домен холодового шока – состоит из пяти b-тяжей с РНП-последовательностью, расположенной в одном из тяжей и структурно очень схож с S1 мотивом [38].
КН мотив – содержит стабильную baabba структуру.[11] Точная роль мотива в РНК-связывании неизвестна.
Мотив DSRM – имеет abbba топологию. Возможный участок связывания находится в петле между b-тяжем и С-концевой a-спиралью [9].
RGG мотив – содержит 20-25 аминокислот и обычно имеется в комбинации с РНК-связывающими мотивами других типов. Он состоит из близкорасположенных поворотов Arg-Gly-Gly (RGG), расположенных среди других, часто ароматических аминокислот [8].
Мотив ARM – состоит из 10-20 аминокислот с большим количеством аргинонов. Встречается в вирусных и фаговых белках.
Домены с «цинковыми пальцами» - содержит последовательности СХ4СХ12НХ3-4Н и соответствующее число ионов цинка, связанные цистеинами и гистидинами [17].
Другие РНК-связывающие мотивы. Существует ряд других консервативных последовательностей, не имеющих гомологии с уже перечисленными мотивами. Например, мотив белка-аттенюатора триптофанового оперона из B.subtilis. [2].
1.2.2. Взаимодействия белков с тРНК
Вопрос о том, как аминоацил-тРНК-синтетазы достигают высокой специфичности в узнавании тРНК, вероятно, был одним из первых серьёзно изучаемых вопросов в проблеме РНК-белковых взаимодействий. Чтобы взаимодействовать со всеми компонентами трансляционного аппарата, все тРНК должны иметь достаточно схожую трёхмерную структуру, что значительно затрудняет для каждой из 20 синтетаз выбор своих изоакцепторных тРНК из пула тРНК клетки. В связи с этим, возможности к дискриминации весьма ограничены и, в основном, сведены к узнаванию нуклеотидов в специфических позициях.
К настоящему времени определены элементы узнавания для 19 из 20 синтетаз. Оказалось, что они неодинаковы для разных синтетаз. Большинство этих ферментов узнаёт одну или более из трёх следующих детерминант:
по меньшей мере, один нуклеотид в антикодоне;
одну или более из последних трёх пар нуклеотидов акцепторного стебля;
так называемое «дискриминаторное» основание между акцепторным стеблем и ССА концом.
Кроме того, по крайней мере, 8 синтетаз используют и другие отличительные признаки.
Элемент узнавания может влиять на специфичность аминоацилирования тРНК, потому что он узнаётся «правильной» синтетазой (позитивный элемент) или потому что предотвращает взаимодействие с «не своей» синтетазой (негативный элемент). Негативный элемент может маскировать присутствие позитивных элементов [32]. Яркой иллюстрацией этого факта являются наборы из трёх элементов специфичности около ССА конца тРНК для аланина, гистидина и глицина. Каждая детерминанта является позитивным элементом для одной или двух синтетаз и негативным для остальных [19].
Несмотря на выполнение одной и той же реакции аминоацилирования, синтетазы могут осуществлять различное связывание с субстратом реакции – тРНК, причём элементы узнавания тРНК не всегда являются уникальными, а могут иметь структурное сходство с другими классами белков.
1.2.3. Взаимодействия белков с матричными РНК
Механизм мРНК-белковых узнаваний является, пожалуй, наименее изученным вопросом в проблеме РНК-белковых взаимодействий, в виду отсутствия структур, решённых с достаточно высоким разрешением (исключение - комплекс L30-мРНК).
Однако, опираясь на данные полученные методом ядерного магнитного резонанса было сделано предположение, что основную роль в процессе мРНК-белкового узнавания играют приведённые ранее консервативные мотивы. Например, RNP1 и RNP2 (белок U1A), домен холодового шока и гидрофильный С-концевой домен с чередующимися кластерами отрицательно и положительно заряженных аминокислотных остатков (Y-box-белки).
1.2.4. Взаимодействия белков с рРНК
Взаимодействия белков с рРНК очень сильно отличаются от взаимодействий белков с ДНК и тРНК. Места связывания белков на ДНК и тРНК характерны наличием специфической эволюционно консервативной последовательности нуклеотидов, замена которых приводит к сильному ослаблению или исчезновению взаимодействий [1]. Предположение об определяющей роли последовательности нуклеотидов в РНК-белковом узнавании, основанное на исследованиях комплексов белков-репрессоров с ДНК и изучении аминоацил-тРНК-синтетаз, оказывается некорректным применительно к рРНК-белковым комплексам. Поскольку структура ДНК регулярна, именно последовательность оснований, а не конформация остова, определяет специфичность связывания. Подобное же можно сказать о комплексах аминоацил-тРНК-синтетаз с соответствующими тРНК, поскольку пространственные структуры тРНК однотипны и специфичность узнавания зависит именно от последовательности оснований в определённых участках молекулы тРНК.
рРНК, в отличие от ДНК и тРНК, содержит много нерегулярных участков, и рибосомные белки могут опознавать специфические конформации, образованные сахарофосфатным остовом таких участков. Места связывания рибосомных белков на рРНК можно разделить на две группы [18]. К первой относятся двухцепочечные спирали длиной до 40 нт, содержащие выпетливания (loops) или выпуклости (bulges), которые или искажают структуру спиралей нормальной А-формы, образуя уникальные конформации, опознаваемые белком, или же сами формируют места связывания. Вторая группа включает домены рРНК, имеющие сложную пространственную структуру, взаимное расположение сегментов которой стабилизируется рибосомными белками.
Для поиска предполагаемых мест РНК-белкового связывания в молекулах рибосомных белков используют две концепции:
Наличие кластеров эволюционно консервативных положительно заряженных или ароматических аминокислотных остатков в структурах белков может служить указанием на функциональную важность данной области молекулы при взаимодействии с рРНК;
Способность рибосомных белков к специфическим перекрестным взаимодействием с рРНК из эволюционно удаленных организмов позволяет использовать сравнение структур гомологичных белков для локализации возможных мест связывания рРНК и говорить о консерватизме пространственной структуры РНК-белкового интерфейса [31].
Дополнительно используют модификации белка генно-инженерными или биохимическими методами, которые заключаются в замене или химической модификации аминокислотных остатков или же в удалении части полипептидной цепи с последующей проверкой константы связывания.
1.3. Современные методы исследования РНК-белковых взаимодействий
1.3.1. Биохимические методы
К биохимическим методам относятся определение констант связывания на фильтрах, подвижность в геле и центрифугирование в градиенте сахарозы.
1.3.1.1. Связывание на фильтрах
Является стандартно используемой методикой для определения РНК-белковых взаимодействий и оценки константы связывания. Принцип данного метода основан на способности нитроцеллюлозных фильтров удерживать белки, а также связанные РНК, пока несвязанные РНК проходят через фильтр. Несмотря на свою концептуальную простоту, метод всё же не является достаточно надёжным и не подходит для изучения некоторых РНК-белковых комплексов: он хорошо работает для сравнительно небольших молекул РНК, большие же молекулы РНК-белковых комплексов часто не удерживаются.
Методика заключается в том, что эквимолярные количества радиоактивной РНК смешиваются с белком в различной концентрации и фильтруются через нитроцеллюлозные фильтры. В идеале при высокой концентрации белка должно удерживаться до 100% РНК, в действительности же этот процент значительно меньше. Кроме того, белковые фракции также могут не удерживаться фильтром. Например, при анализе взаимодействия белка L18 с 5S рРНК было обнаружено, что эффективность связывания комплекса 5SрРНК-L18 составляла 35% при удержании 65% белка L18, и < 5% для свободной 5S рРНК. Тем не менее, точность измерения не зависит непосредственно от эффективности задерживания.
На оценку эффективности связывания влияют многие параметры, и они должны быть оптимизированы для каждого конкретного случая:
Буфер: Повышение концентрации солей уменьшает эффективность связывания. Для большинства комплексов концентрация одновалентных ионов должна составлять около 150 mM, чтобы гарантировать специфичность связываться.
Температура: Низкая температура увеличивает эффективность связывания.
Скорость подачи буфера и условия промывки: скорость подачи буфера должна быть постоянной (одной и той же) в каждом эксперименте. Неспецифическое удержание несвязанного РНК на фильтре можно уменьшать интенсивной промывкой, но зачастую это ведёт и к снижению специфической сорбции в целом. Поэтому для некоторых комплексов применяется промывка небольшим объёмом, в то время как для других комплексов наилучшие результаты получаются при промывке большими объёмами.
Ренатурация РНК: хорошо известно, что разные препараты РНК дают различную эффективность связывания. Тщательная ренатурация уменьшает вариабельность между этими препаратами [23].
1.3.1.2. Подвижность в геле
Оценка подвижности в геле основана на различии в подвижности между свободными РНК и РНК-белковыми комплексами при их миграции в нативном полиакриламидном геле. Возрастающий размер комплекса, а также небольшой положительный заряд связанных с белками нуклеиновых кислот вызывает различие в их подвижности.
Метод позволяет напрямую оценить стехиометрию РНК-белкового комплекса. Если связывается более чем один белок, то видны дополнительные сдвиги (supershifts) в мобильности РНК. При добавления антител, эту особенность можно использовать для идентификации белков в комплексе, когда для образования комплекса используются неизвестные препараты белка.
Основная проблема выполнения данной методики – это выбор условий, обеспечивающих специфическое РНК-белковое связывание. Главное при этом - выдержать высокую концентрацию солей (300-500 mM NaCI или KC1), но при этом надо учитывать, что высокая концентрация соли зачастую увеличивает и процент диссоциации комплекса при электрофорезе. Таким образом, концентрация солей должна быть оптимизирована в зависимости от конкретного эксперимента. Многие эксперименты по определению подвижности в геле РНК-белковых комплексах изучаются в 50 mM трис-глициновых буферах, а, например, специфические комплексы Escherchia coli требуют по крайней мере дополнительного добавления 150 mM Na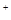 и 5 mM Mg и 5 mM Mg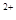 . .
Если комплекс чувствителен к повышению солевой концентрации, неспецифическое связывание может быть уменьшено добавлением носителя РНК [23].
1.3.1.3. Центрифугирование в сахарозном градиенте
Центрифугирование в градиенте сахарозы является наиболее простым, но в тоже время менее чувствительным методом анализа РНК-белковых комплексов, основным достоинством которого являются значительно большие возможности при выборе ионных условий, чем, например, при использовании электрофоретических методов. При применении возможно формирования специальных градиентов, таких как изокинетический градиент для определения коэффициентов седиментации.
Наиболее распространённым типом градиента является линейный градиент сахарозы. Поскольку рибонуклеопротеиновые комплексы более плотные по сравнению с максимально возможной плотностью сахарозы (1.3 г/мл), они всегда образуют осадок в нижней зоне. Так что выбор градиента для разделения определяется зависимостью от количества осаждённой части: 5-20% (w/w) градиент обычно выбирается для разделения малых нуклеопротеинов, 10-30% (w/w) градиент - для разделения сплайсосом, рибосомных субъединиц и моносом, поскольку 20-47% (w/w) градиент - для объектов полисомных размеров [23].
1.3.2. Физические методы
Основаны на решении пространственных структур РНК-белковых комплексов с атомарным разрешением. К этим методам относят метод ядерного магнитного резонанса (ЯМР) и метод рентгеноструктурного анализа.
Метод ЯМР позволяет видеть структуру макромолекулы в растворе без ограничений на взаимное расположение атомов, накладываемых кристаллической упаковкой. В настоящее время из-за сложности и величины РНК-белковых комплексов сфера его применения сильно ограничена.
Наиболее эффективным и универсальным методом определения трёхмерной структуры РНК-белковых комплексов с атомным разрешением является рентгеноструктурный анализ. Метод основан на свойстве биологических макромолекул образовывать монокристаллы, представляющих собой множество идентичных молекул, упорядоченных определенным образом. С кристаллов возможно получение рентгеновской дифракционной картины, с последующим анализом полученных данных и построении на их основе карты электронной плотности. Затем получают пространственную модель структуры, которая после ряда уточнений минимизируется по химической и кинетической энергиям [5].
Наряду с методом ядерного магнитного резонанса, рентгеноструктурный анализ также не лишён своих недостатков, наиболее значимыми из которых являются проблема кристаллизации РНК-белковых комплексов (необходимо наличие достаточно крупных, стабильных и однородных кристаллов), а так же фазовая проблема, решаемая методами изоморфного замещения, молекулярного замещения и аномальной дисперсии на нескольких длинах волн.
В последние годы, благодаря использованию синхротронного излучения, новых типов детекторов, а также разработке новых программ для обработки данных и расчёта фаз, метод рентгеноструктурного анализа получил наиболее широкое распространение.
2. Экспериментальная часть
2.1. Материалы и методы исследования
2.1.1. Метод молекулярного замещения
Для решения проблемы фаз в данной работе использовался метод молекулярного замещения. Метод основан на использовании известной структуры гомологичной молекулы (белка, РНК или ДНК) в качестве модели для получения начального приближения набора фаз [16].
Для расчета начального набора фаз необходимо, чтобы модель наилучшим образом аппроксимировала положения неизвестной молекулы в элементарной ячейке кристалла. Определение таких положений модели является основной задачей метода молекулярного замещения, которая обычно решается в два этапа. На первом этапе определяется ортогональное преобразование W, обеспечивающее правильную ориентацию модели в кристаллической ячейке. На втором этапе проводится поиск вектора трансляции v, задающего положение ориентированной модели в элементарной ячейке кристалла. Чаще всего, вышеупомянутое ортогональное преобразование выражается через углы Эйлера или сферические углы [37].
Функции вращения
Преобразование можно найти путем сравнения функций Паттерсона кристалла неизвестной молекулы Px и модели Pm. Критерием соответствия ориентации модели и неизвестной молекулы служит так называемая функция вращения, которая представляет собой интеграл перекрывания функций Px(r) и Pm(r) в элементе объема U и имеет максимумы если системы внутренних векторов модели и неизвестной молекулы ориентированы одинаково. Существуют методы расчета функции вращения как в прямом [22] так и в обратном пространстве [37]. В случае прямого пространства функции Паттерсона Px и Pm рассчитываются в явном виде при заданном разрешении с помощью преобразования Фурье. Затем происходит вращение Pm относительно Px с заданным шагом и ищутся максимумы функции вращения:
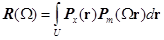 , (1) , (1)
где область интегрирования U - как правило сферический слой с центром в начале координат, задающийся минимальным и максимальным радиусами rmin и rmax, соответственно. Радиус rmin выбирается таким образом, чтобы исключить пик функции Паттерсона в начале координат (обычно rmin ³ 2Å), который может порождать ошибки при численном интегрировании. Радиус rmax выбирается так, чтобы включить в область интегрирования максимальное количество внутримолекулярных векторов при минимально-возможном количестве межмолекулярных [5].
Для уменьшения расчетных затрат при численном интегрировании на сетке используются только те точки, в которых функция Pm принимает наибольшие значения, а значения функции Px в этих точках рассчитываются с помощью процедуры интерполяции [22].
Применяя преобразование Фурье и теорему Парсеваля для уравнения (1) можно получить выражение для функции вращения в обратном пространстве [37]:
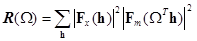 , (2) , (2)
где h = (h,k,l) обозначает миллеровские индексы, а - транспонированную матрицу оператора . Действие на |Fm(h)|2 будет в общем случае приводить к возникновению точек в обратном пространстве, которые не описываются целочисленными индексами (h,k,l). Значения |Fm(h)|2 в таких точках могут быть получены с помощью так называемой интерференционной функции G [2]:
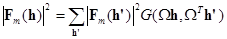 (3) (3)
Анализ максимумов R() позволяет не только выявить наиболее вероятные ориентации модели в ячейке кристалла неизвестной молекулы, но и, в случае нескольких молекул в независимой части элементарной ячейки, найти операции точечной некристаллографической симметрии, связывающие ориентации этих молекул.
2.1.1.2. Функции трансляции
Вторым этапом решения задачи молекулярного замещения является определение положения ориентированной молекулы в ячейке кристалла. Критерием соответствия положения модели и неизвестной молекулы служит функция трансляции. Существует много вариантов определения функции трансляции, в которых используются как функции Паттерсона [3, 36] так и коэффициенты корреляции между экспериментальными и расчетными амплитудами структурного фактора [20]. Функция трансляции может также включать фазовую информацию [4] и ограничения на возможную кристаллическую упаковку молекул [22]. Основной целью при этом является нахождение глобальных максимумов функции трансляции в зависимости от вектора трансляции v, описывающего положение модели в элементарной ячейке. Эта задача обычно решается с помощью процедуры поиска на сетке разбитой по компонентам вектора v.
Функция трансляции, в которой используется перекрывание между экспериментальной и расчитанной по модели функциями Паттерсона имеет общий вид:
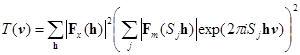 , (4) , (4)
где Sj обозначает операторы симметрии данной группы [15].
Наличие экспериментальной фазовой информации может существенно повысить отношение сигнал-шум в пиках функции трансляции, даже если экспериментальный набор фаз содержит значительные ошибки (например, в тех случаях, когда имеется только одна изоморфная производная). В формулировке Рида и Ширбека [35] функция трансляции, включающая фазовую информацию, определяется следующим образом:
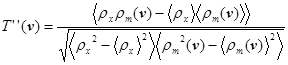 , (5) , (5)
где ρx и ρm – функции экспериментальной и модельной электронной плотности, соответственно.
Кроме правильной ориентации модели, к основным факторам влияющим на точность решения функции трансляции относятся качество и полнота модели и рентгеноструктурных данных, диапазон разрешений, а также критерий отбора в соответствии с которым те или иные структурные амплитуды включаются в расчет. Также как и для функции вращения, исключение слабых рефлексов из экспериментального набора данных (без заметного ущерба для полноты набора) может несколько снизить уровень шума функции трансляции [7].
После того как решения функции трансляции получены их уточняют с помощью процедуры оптимизации ориентации и положения модели как твердого тела по методу сопряженных градиентов (например, процедура FIT в AmoRe [11] или RIGID_BODY в CNS [6]).
2.1.1.3. Методы 6-мерного поиска
При использовании моделей плохого качества (например, в случае низкой гомологии) или моделей описывающих лишь малую часть неизвестной структуры часто возникает ряд проблем, затрудняющих решение задачи молекулярного замещения обычными методами. Значительные ошибки функции вращения, неизбежно возникающие в таких случаях, усугубляют собственные ошибки функции трансляции и приводят либо к полному отсутствию правильных решений, либо к тому, что эти решения оказываются среди максимумов, лежащих на уровне шума и нет достоверных критериев позволяющих однозначно выделить их среди прочих.
Единственным на сегодняшний день общим подходом, позволяющим решать вышеперечисленные проблемы и до определенной степени расширить границы применимости метода молекулярного замещения, является отказ от разделения задачи на поиск решений функций вращения и трансляции и применение процедуры 6-мерного поиска с одновременным варьированием как углов Эйлера (α,β,γ), так и компонент вектора трансляции (vx,vy,vz). Но, несмотря на значительный прогресс вычислительной техники, ни в одной из существующих программ, включая самые современные, 6-мерный поиск не проводится напрямую, как систематический поиск на 6-мерной сетке. Таким образом, ни одна из существующих программ не гарантирует нахождения абсолютных максимумов объединенной функции вращения-трансляции.
Не так давно, двумя группами независимо были предложены стохастические алгоритмы 6-мерного поиска, которые позволили создать программы, ставшие стандартным инструментом в рентгеновской кристаллографии макромолекул:
В работе Киссинджера и др. [26] был применен так называемый эволюционный алгоритм, который принадлежит к семейству алгоритмов стохастической оптимизации, включающему такие методы как Монте-Карло [28] и медленный отжиг [25].
Генетический алгоритм, независимо предложенный Чангом и Льюисом [14], основан на том же самом принципе, что и эволюционный и отличается от последнего лишь некоторыми деталями реализации. Подобный подход применялся также для разработки процедуры поиска положений тяжелых атомов в тяжелоатомных производных кристаллов макромолекул [13] и в прямых методах расчета фаз для кристаллов вирусных частиц [29].
Эволюционный алгоритм использует принцип естественного отбора для нахождения оптимальных решений. Вначале генерируется набор случайных решений, задающих одновременно и ориентацию и положение модели в элементарной ячейке. Затем рассчитываются структурные факторы Fm для каждого решения и производится отбор лучших решений исходя из коэффициента линейной корреляции [26].
Отобранные решения сохраняются и используются для создания нового набора с тем же количеством элементов, что и в предыдущем. Недостающие элементы нового набора получают, внося в ориентации и положения отобранных решений случайные изменения в соответствии с нормальным распределением. Таким образом, плотность распределения элементов нового набора уже не будет равномерной, а будет иметь максимумы в окрестностях отобранных решений. Затем снова происходит расчет структурных факторов Fm, отбор лучших решений, создание следующего набора, и так далее, пока не будет получено решение с некоторым оптимальным значением коэффициента линейной корреляции. На последней стадии, для лучшего отобранного решения проводится оптимизация ориентации и положения модели как твердого тела по методу сопряженных градиентов [33].
Скорость 6-мерного поиска с использованием эволюционного алгоритма значительно увеличивается за счет применения метода непрерывных преобразований структурных факторов [14, 26]. В этом методе структурные факторы рассчитываются один раз с помощью быстрого преобразования Фурье (FFT) для модели, помещенной в начало координат искусственной ячейки симметрии P1. В ходе 6-мерного поиска, изменение ориентации модели учитывается путем ортогональных преобразований индексов обратной решетки и использования линейной интерполяции в обратном пространстве. Изменение положения модели учитывается применением соответствующих фазовых сдвигов. При этом, принимаются во внимание вклады всех симметрически связанных молекул.
Одной из серьезных проблем, возникающих при уточнении фаз, полученных методом молекулярного замещения, является так называемая “model-bias” проблема [34]. Эта проблема проявляется в том, что если часть модели не соответствует реальной структуре, то на карте электронной плотности, рассчитанной по модели, данный участок будет в большей степени соответствовать модели чем реальной структуре. Традиционно, эта проблема решается с помощью OMIT карт электронной плотности, рассчитанных по модели, из которой исключен интересующий участок. С введением Брюнгером в программу CNS [6] возможности модификации электронной плотности рассчитанной по модели, появился еще один способ решения этой проблемы.
Таким образом, метод молекулярного замещения представляет собой мощный инструмент в кристаллографии макромолекул, который в случае высокой гомологии позволяет быстро и эффективно решать фазовую проблему. По мере того как количество известных структур макромолекул неуклонно растет, метод молекулярного замещения становится все более и более актуальным.
2.1.2. Сбор и обработка дифракционных данных
Сбор дифракционных данных комплекса S15-16SрРНК (S15 T3C мутант) проводился на синхротроне ESRF (Гренобль, Франция), линия ID14 С использованием детектора Mar CCD (l= 0.93300 Å; Т=100К).
Полученные данные обрабатывали программами DENZO и SCALEPACK. Обработка данных имеет ряд этапов:
Выделение сильных рефлексов;
Предсказание обратной решётки;
Определение пространственной группы и параметров ячейки;
Уточнение параметров кристалла и детектора;
Обработка всех имеющихся изображений;
Приведение рефлексов к общей шкале;
Интегрирование рефлексов по всему обратному пространству.
В результате мы получили набор дифракционных данных в обратном пространстве.
2.1.3. Решение проблемы фаз
Первоначальный набор фаз получен с помощью метода молекулярного замещения. Основой для модели послужила структура с заменой Met на SeMet комплекса S15(I11M+A79M)-16SрРНК.
Для решения задачи молекулярного замещения использовалась процедура оптимизации ориентации и положения модели как твердого тела по методу сопряженных градиентов. Расчеты проводились с помощью программы комплекса CNS RIGIT-BODY REFINEMENT. Полученный R-фактор (0,339), показал, что при общей гомологии комплексов (»99%) всё же наблюдается расхождение в боковых цепях и петлях.
2.1.4. Построение и уточнение модели
Первоначально были проведены несколько циклов автоматического (программой CNS) кристаллографического уточнения, которое включало в себя молекулярно-динамическую процедуру моделированного отжига (ANNELING), проводимую во внутренних координатах; уточняемыми параметрами были торсионные углы. При этом использовались ограничения (типа “restraints”) в соответствии с операцией некристаллографической симметрии.
Затем были получены две карты электронной плотности:
карта, рассчитанная с использованием коэффициентов 2fo-fc, где fo – структурный фактор, полученный экспериментально на основе дифракционных данных, fc – структурный фактор, рассчитанный на основе модели
composit-omit map – разностная 2fo-fc карта, где fc определяется как комбинированная величина, рассчитанная по нескольким моделям с исключением 5% атомов на каждом шаге.
Полученные карты были хорошего качества, что позволило вписать все боковые остатки белка, уточнить положение главной цепи и молекулы РНК. Для ручной правки использовалась программа молекулярной графики О.
Окончательное уточнение включало уточнение параметров длин связей и углов с помощью программы MINIMIZE (CNS), уточнение В-факторов для индивидуальных атомов, а так же проверку стереохимии полученной структуры и расстояний между атомами (водородные связи, Ван-дер-Вальсовы взаимодействия, «плохие» расстояния).
Окончательная модель, уточненная до значений R-фактора ***8% и Rfree ***% при разрешении до 2,8Е, включает ** аминокислотных остатков и ** нуклеотидов. Кроме того были локализованы *** молекул воды, ** ионов ионов Mg+2 в независимой части элементарной ячейки. Общее число атомов модели в независимой части элементарной ячейки составило ****. Модель обладает хорошими стехиометрическими параметрами и не содержит остатков, расположенных в запрещенных областях карты Рамачандрана. Часть окончательной 2Fo-Fc карты электронной плотности на уровне 1.5 s показана на рис. 20. Структура комплекса S15-16SрРНК схематически показана на рис. 21. Координаты атомов окончательной модели комплекса S15-16SрРНК занесены в RCSB Protein Data Bank (код ****).
2.2. Результаты и их обсуждение
В результате проделанной работы была построена модель структуры комплекса S15-16SрРНК (S15 T3C мутант).
Модель была уточнена до R-фактора 24,5% и Rfree 30,9% при разрешении до 2,9Е, и включает 86 аминокислотных остатков и 57 нуклеотидов. Общее число атомов модели в независимой части элементарной ячейки составило 1943. Модель обладает хорошими стехиометрическими параметрами и не содержит остатков, расположенных в запрещенных областях карты Рамачандрана.
Таблица 2.2.1. Статистика уточнения структуры комплекса комплекса S15-16SрРНК (S15 T3C мутант)
| Пространственная группа |
Р6422 |
| Параметры ячейки (Е) |
a=128.228, b= 128.2286, c=64.9505 |
| Длина волны (Е) |
0,933 |
| Разрешение (Е) |
500 - 2,9 |
| Избыточность |
5,9 |
| Полнота набора (%) |
98,0 |
| Rsym (%) |
3,1 |
| Rcryst/Rfree (%) |
24,5/30,9 |
| Разрешение (Е) |
6,0 – 2,9 |
| Число рефлексов |
8209 |
Вср (Е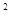 ) ) |
44,8 |
| R.m.s.d. связей (Е) |
0,006 |
| R.m.s.d. углов (°) |
1,2 |
Как видно из рисунка 2.2.1., белок S15 относится к семейству a-спиральных белков и состоит из четырёх a-спиралей, взаимодействующих с 16SрРНК и стабилизирующих данную структуру.
Фрагмент 16S рРНК из T.thermophilus (рис. 2.2.2.) длиной 57 нт состоит из укороченных спиралей Н20, Н21 и Н22. Спираль Н22 стыкуется со спиралью Н21, образуя вытянутую структуру, а спираль Н20 прилегает к ней под углом примерно в 60°.
рис.2.2.1. Структура белка S15 (T3C мутант)
рис. 2.2.2. Структура фрагмента 16S рРНК
Белок S15 распознаёт на поверхности РНК два удалённых участка, один из которых располагается в верхней части спирали H22, а другой – в районе соединения трёх спиралей РНК (рис. 2.2.3.). Участки отстают друг от друга на один виток спирали.
рис.2.2.3. Структура комплекса S15-16SрРНК (S15 T3C мутант)
Выводы:
Получен и обработан набор дифракционных данных для комплекса S15-16SрРНК (S15 T3C мутант).
Методом молекулярного замещения определена структура комплекса.
Показано, что рибосомный белок комплекса S15 связывается с 16S рРНК в двух пространственно удалённых участках.
Показано, что мутация Т3С не изменяет пространственной структуры комплекса и структуры РНК-белкового интерфейса.
Список литературы
Серганов А.А. (1997) Изучение РНК-связывающих свойств рибосомного белка S15 из Thermus thermophilus. Диссертация, МГУ.
Anston A.A., Otridge J., Brzozowski A.M., Dodson E.J., Dodson G.G., Wilson K.S., Smith T.M., Yang M., Kurecki T., Gollnick P. (1995). Biochemestry. 18: 693-700
Argos P. & Rossmann M.G. (1980) in “Theory and Practice of Direct Methods in Crystallography” (Ladd and Palmer, eds.), pp.361-417. Plenum, New York.
Bentley G.A. & Houndusse A. (1992), Acta Cryst. A48, 312-322.
Blundell T.L. & Johnson L.N. (1974) Protein Crystallography. Academic Press, New York.
Brunger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J., Kuszewski J., Nilges M., Pannu N.S., Read R.J., Rice L.M., Simonson T., Warren G.L. (1998) Crystallography & NMR System: A New Software Suite for Macromolecular Structure Determination. Acta Cryst. D54, 909-921.
Brunger A.T., Milburn M.V., Tong L., de Vos A.M., Jancarik J., Yamazumi Z., Nishimura S., Ohtsuka E., Kim S.-H. (1990), Proc. Natl. Acad. Sci. USA 87, 4849-4853.
Burd C.G. Dreifuss G. (1994). Conserved structures and diversity of functions of RNA-binding proteins. Science 265: 615-620
Bycroft M., Grunert S., Murzin A., Proctor M., Johnston D. (1995). NMR solution structure of a dsRNA binding domain from Drosophila staufen protein reveals homology to the N-terminal domain of ribosomal protein S5. EMBO J. 14: 3563-3571
Bycroft M., Hubbard T.J.P., Proctor M., Freund S.M.V., Murzin A. (1997). The solution structure of the S1 RNA binding domain: a member of an ancient nucleic acid-binding fold. Cell 88: 235-242
Castellano E., Oliva G., Navaza J. (1992), J. Appl. Cryst. 25, 281.
Cate J.H., Gooding A.R., Podell E., Zhou K., Golden B.L., Szewczak A.A., Kundrot C.E., CechT.R., Doudna J.A. (1996). RNA tertiary structure mediation by adenosine platforms. Science 273: 1696-1699
Chang G. & Lewis M. (1994), Acta Cryst. D50, 667-674.
Chang G. & Lewis M. (1997) Molecular Replacement Using Genetic Algorithms. Acta Cryst. D53, 279-289.
Crowther R.A. & Blow D.M. (1967), Acta Cryst. 23, 544-548.
Crowther R.A. (1972) in “The Molecular Replacement Method”, Int. Sci. Rev. No.13 (Rossmann M.G., ed.). Gordon & Breach, New York.
Draper D.E. (1995). Protein-RNA recognition. Annu. Rev. Biochem. 64: 593-620
Draper D.E. (1996) Ribosomal protein-RNA interactions. In Ribosomal RNA: structure, evolution, processing and function in protein biosynthesis. Eds. Zimmermann R.A., Dahlberg A.E. Boca Raton, Florida, CRC Press, 171-198
Franklin C., Shi J.P., Shimmel P. (1992). Overlapping nucleotide determinante for specific aminocylation of RNA. Science. 225: 1121-1125
Fujinaga M. & Read R.J. (1987), J. Appl. Cryst. 20, 517-521.
Harada Y., Lifchitz A., Berthou J., Jolles P. (1981), Acta Cryst. A37, 398-406.
Huber R. (1965), Acta Cryst. A19, 353-356.
Kjems J., Egebjerg J., Christiansen J. (1998) Laboratory techniques in biochemistry and molecular biology. Elsevier, 237
Kim J.L., Nikolov D.B., Burley S.K. (1993). Co-crystal structure of TBP recognizing the minor groove of a TATA element. Nature 365: 520-527
Kirkpatrick S., Gelatt C.D. Jr & Vecchi M.P. (1983), Science, 220, 671-680.
Kissinger C.R., Gehlhaar D.K., Fogel D.B. (1999) Rapid automated molecular replacement by evolutionary search. Acta Cryst. D55, 484-491.
Marino J.P., Gregorian R.S., Csankovski G., Grothers D.M. (1995). Ent helix formation between RNA hairpins with complementary loops. Science 268: 1448-1254
Metropolis N., Rosenbluth A.W., Rosenbluth M.N., Teller A. & Teller E. (1953), J. Chem. Phys. 21, 1087-1092.
Miller S.T., Hogle J.M. & Filman D.J. (1996), Acta Cryst. D52, 235-251.
Musco G., stier G., Joseph C., Morelli M.A.C., Nilges M., Gibson T.J., Pastore A.. (1996) Thre-dimensional structure and stability of the KH domain: molecular insight into the fragile X syndrome. Cell 85: 237-245
Nevskaya, N., Tishchenko, S., Nikulin, A., Al-Karadaghi, S., Liljas, A., Ehresmann, B., Ehresmann, C., Garber, M., Nikonov, S. (1998) Crystal Structure of Ribosomal Protein S8 from Thermus Thermophilus Reveals a High Degree of Structural Conservation of a Specific RNA Binding Site. J.Mol.Biol. 279, 233-244.
Normanly J., Abelson J. (1989). TRNA identity. Annu. Rev. Biochem. 58: 1029-1049
Powell M.J.D. (1977), Math. Programming, 12, 241-254.
Read R.J. (1997) Model Phases: Probabilities and Bias. In Methods in Enzymology, 277, 110-128, Academic Press.
Read R.J. & Schierbeek A.J. (1988), J. Appl. Cryst. 21, 490-495.
Rossmann M.G., Blow D.M., Harding M.M., Coller E. (1964), Acta Cryst. 17, 338-342.
Rossmann M.G. & Blow D.M. (1962), Acta Cryst. 15, 24-31
Schindelin H., Marahiel M.A., Heinemann U. (1993). Universal nucleic acid-binding domain revealed by crystal structure og the B.subtilis major cold-shock domain. Nature 364: 164-168
Seeman N., Rosenberg J.M., Rich A. (1976). Sequence-specific recgnition of double helical nucleic acids by proteins. Pros. Natl, Acad. USA 73: 804-808
von Hippel P.H., McGhee J.D. (1972). DNA-protein interactions. Annu. Rev. Biochem. 41: 231-298
Weeks K.M., Crothers D.M. (1993). Major grove accessibility of RNA. Science 261: 1574-1577
|
