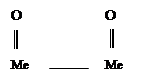Глава 1
Коррозия металла
1. Основы теории коррозии
Термин коррозия происходит от латинского "corrosio", что означает разъедать, разрушать. Этот термин характеризует как процесс разрушения, так и результат.
Среда в которой металл подвергается коррозии (коррозирует) называется коррозионной или агрессивной средой.
В случае с металлами, говоря об их коррозии, имеют ввиду нежелательный процесс взаимодействия металла со средой. Физико-химическая сущность изменений, которые претерпевает металл при коррозии является окисление металла.
Любой коррозионный процесс является многостадийным:
1) Необходим подвод коррозионной среды или отдельных ее компонентов к поверхности металла.
2) Взаимодействие среды с металлом.
3) Полный или частичный отвод продуктов от поверхности металла (в объем жидкости, если среда жидкая).
Известно что большинство металлов ( кроме Ag,Pt,Cu,Au) встречаются в природе в ионном состоянии: оксиды, сульфиды, карбонаты и др., называемые обычно руды металлов.
Ионное состояние более выгодно, оно характеризуется более меньшей внутренней энергией. Это заметно при получение металлов из руд и их коррозии. Поглощенная энергия при восстановлении металла из соединений свидетельствует о том , что свободный металл обладает более высокой энергией, чем металлическое соединение. Это приводит к тому, что металл находящийся в контакте с коррозионно-активной средой стремится перейти в энергетически выгодное состояние с меньшим запасом энергии.
Коррозионный процесс является самопроизвольным, следовательно G=G-G (G и G относятся к начальному и конечному состоянию соответственно). Если G>G то G<0, т.е. коррозионный процесс возможен; G>0 коррозионный процесс невозможен; G=0 система металл-продукт находится в равновесии. То есть можно сказать, что первопричиной коррозии металла является термодинамическая неустойчивость металлов в заданной среде.
1.1 Классификация коррозионных процессов.
1. По механизму процесса различают химическую и электрохимическую коррозию металла.
Химическая коррозия - это взаимодействие металлов с коррозионной средой, при котором окисляется металл и восстанавливается окислительные компоненты коррозионной среды протекают в одном акте. Так протекает окисление большинства металлов в газовых средах содержащих окислитель (например, окисление в воздухе при повышении температуры)
Реклама

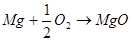 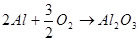
Электрохимическая коррозия - это взаимодействие металла с коррозионной средой, при котором ионизация атомов металла и восстановление окислительной компоненты среды происходит не водном акте, и их скорости зависят от электродного потенциала
металла. По такому процессу протекают, например, взаимодействие металла с кислотами:
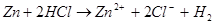
эта суммарная реакция состоит из двух актов:
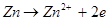 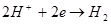
2. По характеру коррозионного разрушения.
Общая или сплошная коррозия при которой коррозирует вся поверхность металла. Она соответственно делится на равномерную (1а), не равномерную (1б) и избирательную (1в), при которой коррозионный процесс распространяется преимущественно по какой-либо структурной составляющей сплава.
Местная коррозия при которой коррозируют определенные участки металла:
а) коррозия язвами - коррозионные разрушения в виде отдельных средних и больших пятен (коррозия латуни в морской воде)
б) межкристаллическая коррозия при ней процесс коррозии распространяется по границе металл-сплав (алюминий сплавляется с хромоникелем) и другие виды коррозии.
3. По условиям протекания процесса.
а) Газовая коррозия - это коррозия в газовой среде при высоких температурах. (жидкий металл, при горячей прокатке, штамповке и др.)
б) Атмосферная коррозия - это коррозия металла в естественной атмосфере или атмосфере цеха (ржавление кровли, коррозия обшивки самолета).
в) Жидкостная коррозия - это коррозия в жидких средах: как в растворах электролитов, так и в растворах не электролитов.
г) Подземная коррозия - это коррозия металла в почве
д) Структурная коррозия - коррозия из-за структурной неоднородности металла.
е) Микробиологическая коррозия - результат действия бактерий
ж) Коррозия внешним током - воздействие внешнего источника тока (анодное или катодное заземление)
з) Коррозия блуждающими токами - прохождение тока по непредусмотренным путям по проекту.
и) Контактная коррозия - сопряжение разнородных электрохимических металлов в электропроводящей среде.
к) Коррозия под напряжением - одновременное воздействие коррозионной среды и механического напряжения.
1.2 Показатель скорости коррозии.
Для установления скорости коррозии металла в данной среде обычно ведут наблюдения за изменением во времени какой-либо характеристики, объективно отражающей изменение свойства металла.
Реклама

Чаще всего в коррозионной практике используют следующие показатели.
1) Показатель изменения массы - изменение массы образца в результате коррозии отнесенный к единице поверхности металла S и к единице времени (например, г/м ч) в зависимости от условий коррозии различают:
а) отрицательный показатель изменения массы
К-
m
=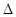 m / S m / S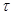  
где m - убыль массы металла за время коррозии после удаления продуктов коррозии.
б) положительный показатель изменения массы
К+
m
=m / S
где m - увеличение массы металла за время вследствие роста пленки продуктов коррозии.
Если состав продуктов коррозии известен, то можно сделать пересчет от К к К и наоборот
К-
m
=К+
m
(nok
A Me / n Me Aok
)
где А и М - атомная и молекулярная масса Ме и окислителя соответственно; n и n валентность металла и окислителя в окислительной среде.
2) Объемный показатель коррозии
К - объем поглощенного или выделившегося в процессе газа V отнесенный к единице поверхности металла и единице времени (например, см/см ч).
К= объ.V/ S
объем газа обычно приводят к нормальным условиям.
Применительно к электрохимической коррозии когда процесс катодной деполяризации осуществляется за счет разряда ионов водорода, например, по схеме 2Н + 2е = Н, или ионизация молекул кислорода О + 4е +2НО = 4ОН; вводятся соответственно кислородный (К ) и водородный (К ) показатель соответственно.
Водородный показатель коррозии - это объем выделившегося Н в процессе коррозии, отнесенный к Su .
Кислородный показатель коррозии - это объем поглощенного в процессе О , отнесенный к Su .
3) Показатель сопротивления.
Изменение электрического сопротивления образца металла за определенное время испытаний также может быть использован в качестве показания коррозии (К).
КR
= (R/Ro
)100% за время t
где Ro
и R электрическое сопротивление образца соответственно до и после коррозии.
У этого способа есть некоторый недостаток толщина металла во все время испытаний должна быть одинаковой и по этой причине чаще всего определяют удельное сопротивление, т.е. изменение электрического сопротивления на единицу площади образца (см, мм) при длине равной единице. Этот метод имеет ограничения применения (для листового металла не более 3мм). Наиболее точные данные получают для проволочных образцов. Этот метод не пригоден для сварных соединений.
4) Механический показатель коррозии.
Изменение какого-либо свойства металла за время коррозии . Сравнительно часто пользуются изменением предела прочности. Прочностной показатель при этом выражается:
Кo
= (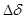 в
/ в
/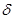 во
) 100% за время t во
) 100% за время t
где o
изменение предела прочности при растяжении после коррозии образца в течении времени ; во
предел прочности до коррозии.
5) Глубинный показатель коррозии.
К - глубина разрушения металла П в единицу времени (например, мм/год)
КП
= п/
Глубина коррозионного разрушения П может быть средней или максимальной. Глубинный показатель коррозии можно использовать для характеристики как равномерной., так и неравномерной коррозии (в том числе и местной) металлов. Он удобен для сравнения скорости коррозии металла с различными плотностями. Переход от массового, токового и объемного к глубинному возможен при равномерной коррозии.
2. Электрохимическая коррозия.
Электрохимическая коррозия является наиболее распространенным типом коррозии металлов. По электрохимическому механизму коррозируют металлы в контакте с растворами электролитов (морская вода, растворы кислот, щелочей, солей) . В обычных атмосферных условиях и в земле металлы коррозируют также по электрохимическому механизму , т.к. на их поверхности имеются капли влаги с растворенными компонентами воздуха и земли. Электрохимическая коррозия является гетерогенным и
многостадийным процессом. Ее причиной является термодинамическая неустойчивость металлов в данной коррозионной среде.
Учение о электрохимической коррозии ставит главный вопрос - вопрос о скорости коррозии и тех факторов, которые влияют на нее. С электрохимической точки зрения коррозия металла это не просто процесс окисления металла, т.к. этот переход должен
сопровождаться сопряженно идущим восстановительным процессом. В результате ионизации освобождаются электроны и роль второго восстановительного процесса состоит в их ассимиляции подходящим окислителем (Д), образующим устойчивое соединение.
Ионизация и процесс ассимиляции электронов каким либо элементом среды (обычно Н ионы или О )представляет собой В отличии химического, электрохимические процессы контролируются (зависят) не только от концентрации реагирующих веществ, но и, главным образом, зависят от потенциала поверхности металла.
Потенциал.
На границе раздела двух разнородных фаз происходит переход заряженных частиц - ионов или электронов из одной фазы в другую, следовательно, возникает разность электрических потенциалов, распределения упорядоченных электрических зарядов, т.е.
образование двойного электрического слоя. Возникновение межфазового скачка потенциала можно объяснить следующими основными причинами; но рассмотрим только те, которые приводят к коррозии металлов, а точнее переход катионов металла из электролита на металл (электродный потенциал) адсорбция анионов электролита на металле (адсорбционный потенциал) возникновение ионно-адсорбционного потенциала за счет одновременной адсорбции поляризуемого атома кислорода и перехода
катионов из металла в электролит.
По известным причинам, абсолютное значение межфазовой разности потенциалов измерить нельзя, эту величину можно измерить относительно другой величины и за точку отсчета принимается стандартный водородный потенциал.
Наличие на межфазовой границе металл-раствор электролита двойного электрического слоя оказывает существенное влияние на процесс, а , в частности, на скорость коррозии металлов. При изменении концентрации (плотности) положительных или отрицательных частиц в растворе или металле может измениться скорость процесса растворения металла. Именно их этих соображений электродный потенциал является одной из важнейших характеристик, определяющих скорость коррозии металла.
2.1 Термодинамика электрохимической коррозии металлов.
Стремлением металлов переходить из металлического состояния в ионное для различных металлов различно. Вероятность такого перехода зависит также от природы коррозионной среды . Такую вероятность можно выразить уменьшением свободной энергии при протекании реакции перехода в заданной среде при определенных условиях.
Но прямой связи между термодинамическим рядом и коррозией металлов нет. Это объясняется тем, что термодинамические данные получены для идеально чистой поверхности металла, в то время как в реальных условиях коррозирующий металл покрыт слоем (пленкой) продуктов взаимодействия металла со средой.
Для расчетов изменения свободной энергии реакции при электрохимической коррозии металла используют величины электродных потенциалов. В соответствии с неравенством процесс электрохимической коррозии возможен, если
GT
= - nET
F < 0
где - э.д.с. гальванического элемента, в котором обратимо осуществляется данный коррозионный процесс, В
- обратный потенциал катодной реакции, В
- обратный потенциал металла в данных условиях.
Следовательно, для электрохимического растворения металла необходимо присутствие в растворе окислителя (деполяризатора, который бы осуществлял катодную реакцию ассимиляции электронов), обратимый окислительно-восстановительный потенциал которого положительнее обратимого потенциала металла в данных условиях.
Катодные процессы при электрохимической коррозии могут осуществляться различными веществами.
1) ионами
2) молекулами
3) оксидами и гидрооксидами (как правило малорастворимыми продуктами коррозии, образованными на поверхности металлов)
4) органическими соединениями
Обратимые окислительно-восстановительные потенциалы катодных
процессов можно рассчитать по уравнениям:
(Vk
)обр
= (Vk
)0
обр
+ (RT/nF) 2,303 lg (ap
ok
/ag
в
)
где (Vk
)обр
= (Vk
)0
обр
стандартный окислительно-восстановительный потенциал при ap
ok
/ag
в
=1,
аu, а - активность (приближенно концентрация окислителя и
восстановителя)
pu, q - стехиометрические коэффициенты окислителя и восстановителя в реакции
В коррозионной практике в качестве окислителей-деполяризаторов, осуществляющих коррозию, выступают ионы водорода и молекулы растворенного в электролите кислорода.
Электродная реакция анодного растворения металла (собственно коррозионные потери металла) в общем случае протекают по схеме Me -> Me + ne
При увеличении активности ионов металла (повышение концентрации ионов металла в растворе), потенциал анода возрастает, что приводит к торможению растворения металла. Понижение активности металла, напротив, способствует растворению
металла. В ходе коррозионного процесса изменяются не только свойства металлической поверхности, но и контактирующего раствора (изменение концентрации отдельных его компонентов). При уменьшении, например, концентрации деполяризатора, у катодной зоны может оказаться, что катодная реакция деполяризации термодинамически невозможна.
2.2 Гомогенные и гетерогенные пути электрохимической коррозии.
Причину коррозии металлов в растворах, не содержащих одноименных ионов, объясняет теория необратимых потенциалов. Эта теория рассматривает поверхность металлов как однородную, гомогенную. Основной и единственной причиной растворения (коррозии) таких металлов является термодинамическая возможность протекания анодного и катодного актов. Скорость растворения (коррозии) будет определяться кинетическими факторами. Но гомогенную поверхность металлов можно рассматривать как предельный случай, который может быть реализован, например, в жидких металлах. (ртуть и амальгамы металлов). Для твердых металлов такое допущение будет ошибочным, хотя бы потому что различные атомы сплава (и чистого металла) занимают различное положение в кристаллической решетке. Наиболее сильное отклонение от гомогенной конструкции будет наблюдаться при наличии в металле инородных включений, интерметаллидов, границ зерен и т.д. В этом случае, разумеется, поверхность является гетерогенной. Установлено, что даже при наличии на поверхности металла неоднородностей в целом поверхность остается эквипотенциальной.
Таким образом неоднородность поверхностей сплава не может являться основной причиной общей коррозии металла. Наиболее существенной в подобных случаях является ионизация растворения анодной составляющей вблизи катодной составляющей, это возможно, если на поверхности металлической конструкции возникают гальванические элементы. Рассмотрим некоторые из них:
а) неоднородность металлической фазы, обусловленная неоднородностью сплава, а также в результате микро и макровключений.
б) неоднородность поверхности металла в следствие наличия границ блоков и зерен кристаллов, выход дислокаций на поверхность, анизотропность кристаллов.
в), г) неоднородность защитных пленок на поверхности за счет микро и макропор пленки (в), за счет неравномерного образования на поверхности вторичных продуктов коррозии (г) и др.
Мы рассмотрели два крайних механизма саморастворения металлов: равномерное растворение идеально гомогенной поверхности и растворения (в основном локальное) микроэлементов при пространственном разделении катодных и анодных зон (процессов).
В общем случае, необходимо считаться с возможностью протекания на анодных участках наряду с основными анодными процессами катодных процессов, на катодных же участках могут протекать с пониженной скоростью анодные процессы растворения.
Можно сделать вывод, что нет оснований противопоставлять "гомогенный" и "гетерогенный" пути протекания коррозионных процессов. Правильнее будет их рассматривать как факторы, взаимно дополняющие друг друга. Основной же причиной коррозии металлов остается по-прежнему термодинамическая вероятность протекания в данных условиях на металле анодных процессов ионизации металла и сопряженного с ним катодного процесса деполяризации.
2.3 Анодные процессы при электрохимической коррозии металлов.
Термодинамические основы.
Для протекания коррозионного процесса существенным является состояние форма соединения , в котором находится катион металла в растворе. Ионизация металла с последующим переходом в раствор простых компонентов металла представляет лишь одно из возможных направлений анодных процессов. Форма их конкретного состояния во многом определяется как природой металла и контактирующей с ним средой , так и направлением и величиной поляризующего тока (или электродного потенциала). Переходя в раствор, коррозирующий металл вступает в связь либо с растворителем, либо с компонентами раствора. При этом могут образовываться простые и комплексные соединения с различной растворимостью и с различной адгезией к поверхности металла. При высоких положительных значениях потенциала на аноде возможен процесс окисления воды с выделением кислорода. В зависимости от того, какие процессы или их сочетания протекают на аноде, они могут в значительной мере (а иногда и полностью) контролировать суммарный процесс коррозии.
2.4
Причины анодного растворения металлов.
Простейшими анодными реакциями являются такие , в результате которых образуются растворимые гидратированные и комплексные катионы,. которые отводятся от анода путем диффузии, миграции (перенос за счет электрического поля) или конвекции.
Полярные молекулы жидкости электростатически взаимодействуют с заряженными ионами, образуют сольватные (в случае воды-гидратные) комплексы. Обладающие значительно меньшим запасом энергии чем ионы в кристаллической решетки металла. Величину этого понижения можно оценить, исходя из соображений предложенных Борном. Полный электрический заряд в вакууме обладает энергией, равной потенциальной энергии. Для определения величины энергии заряда представим, что проводящая сфера радиусом r имеет заряд q. Внесение еще одной части заряда dq в сферу должно быть встречено отталкивающими силами df=qdq/r. Поистине огромное уменьшение энергии иона в водном растворе указывает на устойчивость такого состояния в нем. Таким образом, причиной перехода атомов металла с поверхности и их ионизация является электростатическое взаимодействие (сольватация) ионов металла с полярными молекулами растворителя. Следовательно, схему реакции ионизации в контакте с растворителем правильнее записать в виде:
Me + mHO -> Me + mHO +ne.
2.6 Анодная пассивность металлов.
При значительном торможении анодной реакции ионизации металла скорость коррозионного процесса может понизится на несколько порядков. Такое состояние металла принято называть анодной пассивностью. Пассивность можно определить следующим образом: пассивность - состояние повышенной коррозионной устойчивости металла или сплава (в условиях, когда термодинамически он является реакционно способным), Вызванное преимущественным торможением анодного процесса т.е. может произойти так, что в реальных условиях скорость коррозии "активных" элементов оказывается весьма незначительной в следствии наступления пассивного состояния. Например, титан расположенный левее цинка, и хром, расположенный рядом с цинком, в следствии наступления пассивности оказываются более коррозионностойкими в большинстве водных сред, чем цинк. На склонность к пассивному состоянию влияет природа системы металл-раствор. Наибольшую склонность к переходу в пассивное состояние проявляют Ti,Ni,Al,Mg,Fe,Co и др.
Наступление пассивного состояния приводит к значительному изменению формы анодной поляризационной кривой. Кривая может быть разбита на несколько характерных участков:
Вначале скорость анодного растворения металлов возрастает в соответствии с уравнением Тафеля ( =a + blgi)-участок АВ.
Но начиная с В становится возможным процесс образования защитного слоя (фазового или адсорбционного), скорость которого растет при смещении потенциала в положительную сторону. Это приводит к торможению анодного растворения (BD). В точке D, соответствующей потенциалу ( потенциал начала пассивации) скорость образования защитного слоя равна скорости его растворения. Далее идет рост защитного слоя, экранирующего поверхность, скорость анодного растворения резко понижается (DE). В точке Е, соответствующей потенциалу полной пассивации металл оказывается в пассивном состоянии. На участке EF (область пассивного состояния) скорость анодного процесса не зависит от потенциала, а определяется скоростью химического растворения защитной пленки. Ток соответствующий области пассивного состояния, называется током пассивного состояния (i ). Положительнее F возможна ( -потенциал перепассивации) новая ветвь активного растворения с образованием катионов более высокой валентности.
При высоких положительных потенциалах возможен локализованный пробой оксидной пленки - металл начинает растворятся по типу питтинга (PP') называют потенциалом питтингообразования.
Металл запассивированный в данной среде, может сохраняться в пассивном состоянии некоторое время в непассивирующей среде.
3. Депомеризация.
При наличии в растворе газообразного кислорода и не возможностью протекания процесса коррозии с водородной деполяризацией основную роль деполяризатора исполняет кислород коррозионные процессы, у которых катодная деполяризация
осуществляется растворенным в электролите кислородом, называют процессами коррозии металлов с кислородной деполяризацией. Это наиболее распространенный тип коррозии металла в воде, в нейтральных и даже в слабокислых солевых растворах, в морской воде, в земле, в атмосфере воздуха.
Общая схема кислородной деполяризации сводится к восстановлению молекулярного кислорода до иона гидроокисла:
O + 4e +2HO -> 4OH
3.1 Термодинамические возможности кислородной деполяризации.
Протекание процесса коррозии металла с кислородной деполяризацией согласно уравнения возможно при условии:
V(Me)обр
< (VO
2
)обр
где (VO
2
)обр
- обратимый потенциал кислородного электрода,
равный: (VO
2
)0
обр
+ (RT/4F)2,303 lg(PO2
/OH)
Из последнего уравнения следует, что ( ) зависит от рН среды (а ) и парциального давления кислорода.
Значение обратимых потенциалов кислородного электрода при
различных рН среды и Р
| P (атм)
|
V ,B, при рН среды
|
| рН=0
|
рН=7
|
рН=14
|
| 0,21 |
+1,218 |
+0,805 |
+0,381 |
| 1 |
+1,229 |
+0,815 |
+0,400 |
Коррозия металла с кислородной деполяризацией в большинстве практических случаев происходит в электролитах, соприкасающихся с атмосферой, парциальное давление кислорода в которой Р=0,21 атм. Следовательно, при определении термодинамической возможности протекания коррозионного процесса с кислородной деполяризацией следует производить учитывая реальное парциальное давление кислорода в воздухе (см. табл.). Т.к. значения (V ) очень положительны, то условия соблюдаются в очень многих случаях. В следующей таблице приведены значения ЭДС и изменения изобарно-изотермических потенциалов коррозионных процессов с кислородной деполяризацией:
Me + n/2HO + n/4O = Me(OH)
| Металлы
|
Твердый продукт
(
E
)обр
= (
VO
2
)-(
VMe
)обр
|
G
|
| коррозии
|
(
VO2
)
-(VMe)
обр
|
  Mg Mg(OH) +3,104 -71,6 Mg Mg(OH) +3,104 -71,6
Mn MnO +2,488 -25,6
Zn Zn(OH) +1,636 -37,7
Fe Fe(OH) +1,268 -29,3
Fe Fe(OH) +1,164 -26,3
Cu CuO +0,648 -17,3
Cu Cu(OH) +0,615 -14,2
Ag AgO +0,047 -1,1
|
Сопоставляя эти данные с данными по водороду
 Р (атм) рН=0 рН=7 рН=14

5*10 +0,186 -0,288 -0,642
1 0,000 -0,414 -0,828
позволяет указать на, то что кислородная деполяризация более термодинамически возможна чем водородная деполяризация.
Изучение восстановления кислорода на неблагородных металлах (а именно они представляют наибольший интерес с точки зрения коррозии) затрудняется тем, что при катодной поляризации электрода металл может иметь потенциал более положительный, чем равновесный и, следовательно, подвергается окислению (ионизации).
При катодной поляризации в определенном интервале потенциалов будут происходить одновременно два процесса восстановление кислорода и окисление металла. Окисление металла прекратится когда потенциал металла будет равен или станет отрицательнее равновесного потенциала металла. Эти обстоятельства затрудняют изучение процессов кислородной деполяризации.
Схема кислородной деполяризации.
Каждый процесс с кислородной деполяризацией включает следующие последовательные стадии:
1) Растворение кислорода воздуха в растворе электролита.
2) Транспортировка растворенного кислорода в растворе электролита (за счет диффузии или перемешивания) к слою Прандтля.
3) Перенос кислорода в части слоя Прандтля П( )в результате движения электролита.
4) Перенос кислорода в диффузионном слое электролита толщиной или в пленке продуктов коррозии металла к катодным участкам поверхности.
5) Ионизация кислорода:
а) в нейтральных и щелочных растворах
O2
+ 4e + 2 H2
O = 4OH-
(
водн
)
б) в кислых растворах
O2
+ 4e + 4 H+
(водн)
= 2Н2
O
6) Диффузионный или конвектный перенос ионов ОН от катодных участков поверхности корродирующего металла в глубь электролита.
В реальных условиях коррозии металла наиболее затрудненными стадиями процесса являются:
а) реакция ионизации кислорода на катоде. Возникающую при этом поляризацию называют перенапряжением кислорода. Говорят, что процесс идет с кинетическим контролем.
б) Диффузия кислорода к катоду, либо перенапряжение диффузии. В этом случае, говорят, что процесс идет с диффузионным контролем.
Возможны случаи когда обе стадии - ионизация кислорода и диффузия кислорода оказывают влияние на процесс. Тогда говорят, о кинетически-диффузионном контроле.
3.2 Перенапряжение ионизации кислорода.
Перенапряжение ионизации кислорода чаще всего появляется в сильно перемешанных растворах, при интенсивной аэрации раствора (баротаж воздуха и др.), при наличии на металле тонкой пленки электролита (влаги) как и в случае с любой другой катодной реакцией восстановление перенапряжение ионизации кислорода зависит от катодной плотности тока, материала катода, температуры и некоторых других факторов.
Если плотность тока достаточно высока i> А/м то перенапряжение ионизации кислорода является линейной функцией lgi т.е. имеет место зависимость тапа уравнения Тафеля
V = - (Vk
)э
=
х
= a+b lg ik
где а - постоянная зависящая от молярности катода его состояния, Т и пр., численно а=h при i=1; b постоянная зависящая от механизма возникновения перенапряжения. При заторможенности только реакции взаимодействия кислорода с электроном
b=(RT/BnF)n 2,303 = 0,118/ n
Зависимость перенапряжения ионизации кислорода на металлах в растворе: 0,5NaCl + 0,005MNaCO + 0,005MNaHCO (pH=9,2) в атмосфере кислорода при 20 С, раствор перемешивался а) в координатах б) в координатах .
Катодная реакция ионизации кислорода состоит из цепи последовательных элементарных реакций, т.е. протекает стадийно:
а) образование молекулярного иона кислорода
O2
+e = O2
-
б) образование пергидроксила
O2
-
+ H+
= HO2
в) образование пергидроксила иона
HO2
+ e = HO2
-
г) образование перекиси водорода.
HO2
-
+ H+
= H2
O2
д) восстановление перекиси водорода до гидроксил иона и гидроксил-радикала
H2
O2
+e = OH-
+ OH
е) Восстановление гидроксил-радикала до гидроксил иона
OH + e = OH-
Для ряде металлов (Fe,Cu,Au,Pt) при 25 С const b=0.10..0.13.
Это свидетельствует о том, что причиной перенапряжения ионизации кислорода является замедленность элементарной реакции ассимиляции одного электрона (n=1). Для кислых растворов такой реакцией является, по видимому, образование молекулярного иона кислорода
(а), а для щелочных сред - образование пергидроксил-иона (в).
Глава 2
Электрохимические методы защиты металлов от коррозии.
В зависимости от характера коррозии и условий ее протекания применяются различные методы защиты. Выбор того или иного способа определяется его эффективностью в данном конкретном случае, а также экономической целесообразностью. Любой метод защиты изменяет ход коррозионного процесса, либо уменьшая скорость, либо прекращая его полностью. Коррозионные диаграммы, наиболее полно характеризующие коррозионный процесс, должны отражать и те изменения в ходе протекания, какие наблюдаются в условиях защиты. Коррозионные диаграммы можно использовать, поэтому при разработке возможных путей предохранения металлов от коррозии. Они служат основой для выяснения принципиальных особенностей того или иного метода. В связи с этим при рассмотрении существующих методов защиты поляризационные диаграммы будут использованы в их несколько упрощенном виде (4). На таких диаграммах постулируется линейная зависимость между плотностью и потенциалом каждой частной реакции. Это упрощение оказывается вполне допустимым при качественной оценке особенностей большинства методов
Эффективность защиты выражают через коэффициент торможения γ или степень защиты Z
. Коэффициент торможения показывает, во сколько раз уменьшается скорость коррозии в результате применения данного способа защиты
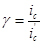
где 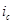 и
и 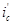 - скорость коррозии до и после защиты. Степень защиты указывает, насколько полно удалось подавить коррозию благодаря применению этого метода: - скорость коррозии до и после защиты. Степень защиты указывает, насколько полно удалось подавить коррозию благодаря применению этого метода:
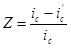
или

2 Катодная защита
Из всех методов защиты основанных на изменении электрохимических свойств металла под действием поляризующего тока, наибольшее распространение получила защита металлов при наложении на них катодной поляризации (катодная защита). При смещении потенциала металла в сторону более электроотрицательных значений (по сравнению с величиной стационарного потенциала коррозии) скорость катодной реакции увеличивается, а скорость анодной падает (см. рис. 1). Если при стационарном потенциале 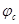 соблюдалосьравенство соблюдалосьравенство
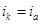 ,
,
то при более отрицательном значении 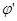 это равенство нарушается: это равенство нарушается:
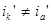
причем
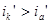 .
.
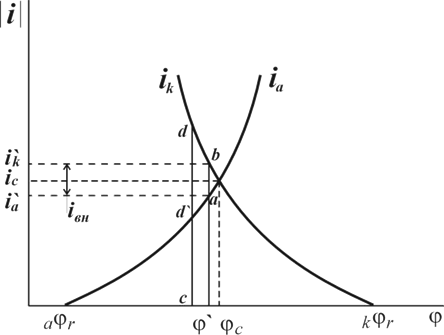
Рис. 1. Поляризационная диаграмма коррозионного процесса.
Уменьшение скорости анодной реакции при катодной поляризации эквивалентно уменьшению скорости коррозии. Коэффициент торможения при выбранном потенциале j/
(см.рис.4) будет равен двум
=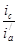 = =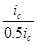 =2, =2,
а степень защиты достигает 50%
=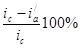 = =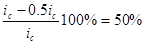 . .
Внешний ток 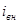 , необходимый для смещения потенциала до значения , представляет собой разницу между катодным и анодным токами
, необходимый для смещения потенциала до значения , представляет собой разницу между катодным и анодным токами
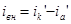
(его величина на рис.4 выражена прямой ав
). По мере увеличения внешнего тока потенциал смещается в более отрицательную сторону, и скорость коррозии должна непрерывно падать. Когда потенциал корродирующего металла достигает равновесного потенциала анодного процесса 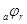 , скорость коррозии делается равной нулю ( , скорость коррозии делается равной нулю (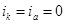 ), коэффициент торможения – бесконечности, а степень защиты 100%. Плотность тока, обеспечивающая полную катодную защиту, называется защитным током
), коэффициент торможения – бесконечности, а степень защиты 100%. Плотность тока, обеспечивающая полную катодную защиту, называется защитным током 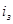 .Его величине на рис.4 соответствует отрезок cd
. Величина защитного тока не зависит от особенностей протекания данной анодной реакции, в частности от величины сопровождающей ее поляризации, а целиком определяется катодной поляризационной кривой. Так, например, при переходе от водородной деполяризации к кислородной сила защитного тока уменьшается и становится равной предельному диффузному току (отрезок cd /
на рис.4).
.Его величине на рис.4 соответствует отрезок cd
. Величина защитного тока не зависит от особенностей протекания данной анодной реакции, в частности от величины сопровождающей ее поляризации, а целиком определяется катодной поляризационной кривой. Так, например, при переходе от водородной деполяризации к кислородной сила защитного тока уменьшается и становится равной предельному диффузному току (отрезок cd /
на рис.4).
Защита металла катодной поляризацией применяется для повышения стойкости металлических сооружений в условиях подземной (почвенной) и морской коррозии, а также при контакте металлов с агрессивными химическими средами. Она является экономически оправданной в тех случаях, когда коррозионная среда обладает достаточной электропроводностью, и потери напряжения (связанные с протеканием защитного тока), а следовательно, и расход электроэнергии сравнительно невелик. Катодная поляризация защищаемого металла достигается либо наложением тока от внешнего источника (катодная защита), либо созданием макрогальванической пары с менее благородным металлом (обычно применяются алюминий, магний, цинк и их сплавы). Он играет здесь роль анода и растворяется со скоростью, достаточной для создания в системе электрического тока необходимой силы (протекторная защита). Растворимый анод при протекторной защите часто называют “жертвенным анодом”.
Катодная защита обычно связана с защитой черных металлов, так как из них изготавливается подавляющая часть объектов работающих под землей и при погружении в воду, например трубопроводы, свайные основания, пирсы, эстакады, суда и др. В качестве материала для расходуемых анодов-протекторов во всем мире широко применяется магний. Обычно он используется в виде сплавов с содержанием 6% алюминия, 3% цинка и 0,2% марганца; эти добавки предотвращают образование пленок, которые снижают скорость растворения металла. Выход защитного тока всегда меньше 100%, так как магний корродирует и на нем выделяется водород. Применяется также алюминий, легированный 5% цинка, но разность потенциалов с железом для сплава значительно меньше, чем для магниевого сплава. Она близка к разности потенциалов для металлического цинка, который также применяется для защиты при условии, что путем соответствующего легирования на анодах предотвращается пленкообразование, связанное с обычным для цинка загрязнением примесями железа Выбор материала для анодов - сложная задача. В почвах или других средах низкой проводимости необходима большая разность потенциалов, поскольку падение iR
между электродами весьма велико, в то время как в средах высокой проводимости возможна более экономичная для использования малая разность потенциалов. Важными переменными являются расположение электродов, рассеивающая способность среды, т. е. ее способность обеспечить одинаковую плотность тока на всех участках защищаемой поверхности, а также поляризационные характеристики электродов. Если электроды погружены в почву, которая по каким – либо причинам неприемлема, например агрессивна по отношению к анодам, то обычно практикуется окружать последние ложем из нейтрального пористого проводящего материала, называемого засыпкой.
Применение для катодной защиты метода приложения тока облегчает регулирование системы и часто дешевле, чем использование анодов – протекторов, которые, конечно, нуждаются в регулярных заменах.
На практике катодная защита редко применяется без дополнительных мероприятий. Требуемый для полной защиты ток обычно бывает чрезмерно велик, и помимо дорогостоящих электрических установок для его обеспечения следует иметь в виду, что такой ток часто будет вызывать вредный побочный эффект, например чрезмерное защелачивание. Поэтому катодная защита применяется в сочетании с некоторыми видами покрытий. Требуемый при этом ток мал и служит только для защиты обнаженных участков поверхности металла.
Давно известно, что скорость коррозии многих металлов часто значительно меньше в растворах сильных окислителей, чем в растворах окислителей более слабых. Сюда относятся такие металлы, как железо, хром, никель, титан, цирконий, алюминий и многие другие. Резкое уменьшение скорости коррозии (на несколько порядков) в сильных окислителях, казалось бы противоречащее термодинамическим свойствам металла и окислителя, называется пассивацией, а состояние металла – пассивным.
Некоторые металлы находятся в пассивном (или близком к пассивному) состоянии даже в таких слабых окислителях, как вода. Это дает возможность практически использовать в качестве конструкционных материалов магний, титан, алюминий и многие другие.
М.В. Ломоносов был, по-видимому, первым исследователем, обратившим внимание на пассивность железа в концентрированной азотной кислоте. Железу посвящены наблюдения М. Фарадея и Х. Шенбейна. М. Фарадей сделал удивительную по научной дальновидности попытку объяснить пассивность железа образованием на поверхности слоя (пленки) окисла или же существованием поверхностных атомов металла в таком состоянии, которое равноценно окислению.
Очень большое практическое значение пассивности, часто определяющее возможность получения сплавов, химически стойких в агрессивных средах, вызвало огромное количество исследований, посвященных изучению пассивного состояния. Если отбросить некоторые несущественные различия, высказываемые на основании сопоставления экспериментальных данных, сказав, что пассивное состояние обусловлено образованием очень тонкой пленки окисла, представляющего собой отдельную фазу, или слоя хемисорбированного кислорода, а может быть и других частиц. Ограничимся представлением о некотором кислородном «барьере», образующемся на поверхности металла в подходящем окислителе и сильно тормозящем анодный процесс.
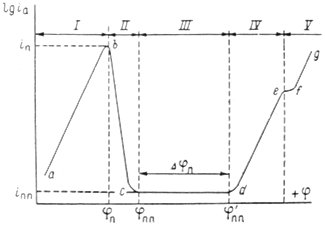
Рис. 2. Полная анодная поляризационная кривая: I – активное растворение; II – переход в пассивное состояние; III – пассивность; IV – перепассивация; V – выделение кислорода.
На рис.5 приведена анодная кривая, которую мы подробно рассмотрим. Активное растворение продолжается до потенциала, отвечающего точке b
. При этом ток равен критическому току пассивации 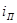 , а потенциал обозначен символом , а потенциал обозначен символом 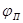 .На участке bс
(в интервале потенциалов - .На участке bс
(в интервале потенциалов -
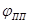 ) происходит пассивация. и ) происходит пассивация. и 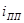 можно назватьпотенциалом и током полной (наилучшей) запассивированности. Это название не вполне точно, так как на реальных поляризационных кривых линия cd
редко бывает строго горизонтальной; но сравнительно небольшими вариациями тока в интервале cd
часто можно пренебречь. Потенциал и ток являются важными характеристиками электрода, показывающими, насколько легко металл переходит в пассивное состояние. Чем отрицательнее и чем меньше , тем легче наступает пассивность. Интервал потенциалов можно назватьпотенциалом и током полной (наилучшей) запассивированности. Это название не вполне точно, так как на реальных поляризационных кривых линия cd
редко бывает строго горизонтальной; но сравнительно небольшими вариациями тока в интервале cd
часто можно пренебречь. Потенциал и ток являются важными характеристиками электрода, показывающими, насколько легко металл переходит в пассивное состояние. Чем отрицательнее и чем меньше , тем легче наступает пассивность. Интервал потенциалов 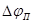 отвечает условиям, в которых сохраняется пассивное состояние. Чем больше , тем в более широких пределах изменение потенциала будет сохранять пассивное состояние. Выше потенциала отвечает условиям, в которых сохраняется пассивное состояние. Чем больше , тем в более широких пределах изменение потенциала будет сохранять пассивное состояние. Выше потенциала 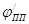 скорость окисления снова увеличивается ( участок cе
) и металл оказывается в области перепассивации или в транспассивном состоянии. Иными словами, отклонение значения потенциала от равновесного значения называется перепассивацией. При еще более высоком потенциале становится возможным процесс окисления ионов гидроксила и выделение кислорода: 4ОН-
→ 2Н2
О + О2
+ 4е
. Это соответствует участку fg
. Если этот последний процесс определяет кинетику анодной реакции, то угловой коэффициент прямой fg
соответствует окислению ионов ОН-
. скорость окисления снова увеличивается ( участок cе
) и металл оказывается в области перепассивации или в транспассивном состоянии. Иными словами, отклонение значения потенциала от равновесного значения называется перепассивацией. При еще более высоком потенциале становится возможным процесс окисления ионов гидроксила и выделение кислорода: 4ОН-
→ 2Н2
О + О2
+ 4е
. Это соответствует участку fg
. Если этот последний процесс определяет кинетику анодной реакции, то угловой коэффициент прямой fg
соответствует окислению ионов ОН-
.
При изменение потенциала в обратном направлении кривая, вообще говоря, имеет такой же ход. В таком случае будет потенциалом начала потери пассивности (депассивации). Его иногда называют фладе-потенциало (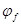 ) по имени исследователя, изучавшего депассивацию железа. ) по имени исследователя, изучавшего депассивацию железа.
В настоящее время вопрос сводится к изучению природы защитных образований на металле, в которых обычно участвует кислород.
При более высоком потенциале становится возможным процесс выделения кислорода:
2Н2
О – 4е
= 2Оадс
+ 4Н+
О2
= 2Оадс
Между адсорбированным кислородом и металлом возникают химические связи (хемисорбция), иначе говоря на поверхности металла возникает хемисорбированная пленка:Из-за неоднородности поверхности пленка может где-то образовываться, а где-то нет. Там, где есть пленка, не происходит окисление металла.
Многие металлы находятся в пассивном состоянии в некоторых агрессивных средах. Хром, никель, титан, цирконий легко переходят в пассивное состояние и устойчиво его сохраняют. Часто легирование металла, менее склонного к пассивации, металлом, пассивирующем легче, приводит к образованию достаточно хорошо пассивирующихся сплавов. Примером могут служить разновидности сплавов FeCr, представляющие собой различные нержавеющие и кислотоупорные стали, стойкие, например, в пресной воде, атмосфере, азотной кислоте и т.д. Для практического использования пассивности нужно такое сочетание свойств металла и среды, при котором последняя обеспечивает значение стационарного потенциала, лежащего в области . Подобное использование пассивности в технике защиты от коррозии известно давно и имеет огромное практическое значение.
Но в последнее время возникло новое направление защиты металлов в таких окислителях, которые сами по себе не способны вызывать пассивность. Известно, что смещение потенциала активного металла в отрицательную сторону должно уменьшить скорость коррозии. Если потенциал становится отрицательнее равновесного в данной среде, то скорость коррозии должна стать равной нулю (катодная защита, применение протекторов). Очевидно, что подобным же образом, но за счет анодной поляризации от внешнего источника электрической энергии можно перевести способный к этому металл в пассивное состояние и тем уменьшить скорость коррозии на несколько порядков. Расход электрической энергии не должен быть велик, так как сила тока в области вообще весьма мала.
Существуют требования, которым должна удовлетворять система, чтобы к ней можно было применить анодную защиту. Прежде всего, нужно надежно знать анодную поляризационную кривую для выбранного металла в данной агрессивной среде. Чем выше , тем большая сила тока потребуется для перевода металла в пассивное состояние; чем меньше , тем меньший расход энергии потребуется для поддержания пассивности; чем шире диапазон , тем большие колебания потенциала можно допустить, т.е. тем легче поддерживать металл в пассивном состоянии. Нужна уверенность в том, что в области металл корродирует равномерно. В противном случае, даже при малой величине , возможно образование язв и сквозного разъедания стенки изделия. Форма защищаемой поверхности может быть довольно сложной, что затрудняет поддержание одинакового значения потенциала на всей поверхности; в этом отношении большая величина особенно желательна. Конечно, требуется и достаточно хорошая электропроводность среды.
Применение анодной защиты целесообразно в сильно агрессивных средах, например в химической промышленности. При наличие поверхности раздела жидкость-газ необходимо иметь в виду, что анодная защита не может распространяться на поверхность металла в газовой среде, что впрочем типично и для катодной зашиты. Если газовая фаза тоже агрессивна или имеется неспокойная поверхность раздела, что приводит к разбрызгиванию жидкости и оседанию капель ее на металл выше поверхности раздела, если происходит периодическое смачивание стенки изделия в определенной зоне, то приходится ставить вопрос об иных способах защиты поверхности выше постоянного уровня жидкости.
Анодная защита может осуществляться несколькими способами.
1. Простое наложение постоянной э.д.с. от постороннего источника электрической энергии. Положительный полюс подключается к защищаемому изделию, а около его поверхности помещают катоды сравнительно малого размера. Они размещаются в таком количестве и на таком расстоянии от защищаемой поверхности, чтобы обеспечить по возможности равномерную анодную поляризацию изделия. Этот способ применяется в том случае, если достаточно велик и нет опасности, при некоторой неизбежной неравномерности распределения потенциала анода, активации или перепассивации, т.е. выхода за пределы .
Таким способом можно защищать изделия из титана или циркония в серной кислоте. Нужно только помнить, что для пассивации сначала потребуется пропускание тока большей силы, что связано с переводом потенциала за . Для начального периода целесообразно иметь дополнительный источник энергии. Следует учитывать также большую поляризацию катодов, плотность тока на которых велика вследствие их малых размеров. Однако, если область пассивного состояния велика, то изменение потенциала катода даже на несколько десятых вольта не представляет опасности.
Периодическое включение и выключение тока защиты, когда изделие уже занассивировано. При включение анодного тока потенциал изделия смещается в отрицательную сторону, причем может произойти депассивация. Но поскольку иногда это происходит довольно медленно, простая автоматика может обеспечить включение и выключение защитного тока в нужное время. Когда потенциал дойдет до величины , т.е. до начала перепассивации, ток выключается; когда потенциал сдвинется в отрицательную сторону до (начало активации), ток снова включается. Смещение потенциала в катодную сторону происходит тем медленнее, чем меньше . Чем ближе был потенциал к величине , тем медленнее он смещается в отрицательную сторону (в направлении ) при выключении тока. Например, для хрома в 0,1н. растворе H2
SO4
при 750
С, если выключение тока произошло при  =0,35 в
, активация наступит через 2 ч
; выключение тока при =0,6 в
вызывает активацию через 5 ч
; выключение же при =1,05в
увеличивает срок начала активации более чем до 127 ч
. Столь большое время, необходимое для депассивации, позволяет делать значительные перерывы в подаче тока. Тогда одной и той же установкой можно обслужить несколько объектов. =0,35 в
, активация наступит через 2 ч
; выключение тока при =0,6 в
вызывает активацию через 5 ч
; выключение же при =1,05в
увеличивает срок начала активации более чем до 127 ч
. Столь большое время, необходимое для депассивации, позволяет делать значительные перерывы в подаче тока. Тогда одной и той же установкой можно обслужить несколько объектов.
Зависимость времени запассивации от потенциала включения легко объяснима при помощи концепции фазового окисла (образуется более толстый слой окисла, растворение которого требует больше времени). Труднее объяснить это явление десорбцией пассивирующего кислорода. Конечно, с ростом положительного значения потенциала прочность связи в адсорбционном слое должна увеличиваться. Но при включении тока разряд двойного слоя происходит сравнительно быстро, хотя адсорбционный слой, возможно, сохраняется долго.
3. Если область пассивного состояния () мала, то необходимо применение потенциостата, поддерживающего заданную величину потенциала (относительно некоторого электрода сравнения) в узких границах. Потенциостат должен быть способен давать большую силу тока.
В настоящее время уже имеется ряд установок для анодной защиты, осуществленных в промышленном масштабе. Защищаются изделия и из обычной углеродистой стали. При анодной защите не только увеличивается срок службы аппаратуры, но также уменьшается загрязнение агрессивной среды продуктами коррозии. Например, в олеуме углеродистая сталь корродирует очень медленно и в этом смысле не нуждается в защите. Но в сосудах для хранения этого продукта происходит загрязнение его железом. Так, без анодной защиты в одной из промышленных установок содержание железа в олеуме составляло ≈ 0,12 %. После наложения защиты концентрация железа снизилась до ≈ 0,004 %, что соответствует его содержанию в исходном продукте. Загрязнение продуктов химической промышленности примесями соединений металлов, являющееся следствием коррозии аппаратуры, во многих случаях весьма нежелательно и даже недопустимо.
Однако, использование анодной защиты связано со значительными трудностями. В то время как катодная защита может употребляться для защиты многих металлов, погруженных в любую электропроводящую среду, например твердую или жидкую, анодная защита применяется только для защиты целых секций химических установок, которые изготовлены из металла, способного пассивироваться в рабочей среде. Именно это и ограничивает ее применение. Кроме того, анодная защита потенциально опасна, поскольку при перерывах подачи тока без немедленного восстановления защиты на рассматриваемом участке начнется очень быстрое растворение, так как разрыв в пленке образует путь с низким сопротивлением в условиях анодной поляризации металла.
Использование анодной защиты требует тщательного проектирования химической установки. Последняя должна иметь такую систему контроля, чтобы любая потеря защиты немедленно привлекала внимание оператора. Для этого может быть достаточным только локальное повышение анодного тока, однако в наихудшем случае может потребоваться немедленное опорожнение всей установки.
Анодная защита не обеспечивает стойкости в присутствии агрессивных ионов. Так, хлоридные ионы разрушают пассивную пленку, а потому их концентрация должна поддерживаться низкой, за исключением защиты титана, который может пассивироваться в хлористоводородной кислоте. В условиях анодной защиты имеет место хорошая рассеивающая способность электролитов и поэтому для поддержания ее установленной защиты требуется сравнительно небольшое количество электродов. Однако при проектировании установок анодной защиты следует учитывать, что в условиях, предшествующих пассивации, рассеивающая способность хуже.
Анодная защита потребляет очень мало энергии и может применяться для защиты обычных конструкционных металлов, способных пассивироваться, например углеродистой и нержавеющей стали, во многих средах. Эта защита легко подвергается контролю и измерениям и не требует дорогостоящей обработки поверхности металла, так как использует самопроизвольный эффект реакции между стенками емкостей и их содержимым. Способ изящен, и его применение, по-видимому, будет расширяться, как только будут преодолены сложности измерения и контроля.
Защита металлов, основанная на изменение их свойств, осуществляется или специальной обработкой их поверхности, или легированием. Обработка поверхности металла с целью уменьшения коррозии проводится одним из следующих способов: покрытием металла поверхностными пассивирующими пленками из его трудно растворимых соединений (окислы, фосфаты, сульфаты, вольфраматы или их комбинации), созданием защитных слоев из смазок, битумов, красок, эмалей и т.п. и нанесением покрытий из других металлов, более стойких в данных конкретных условиях, чем защищаемый металл (лужение, цинкование, меднение, никелирование, хромирование, свинцование, родирование и т.д.).
Защитное действие большинства поверхностных пленок можно отнести за счет вызванной ими механической изоляции металла от окружающей среды. По теории локальных элементов, их эффект следует рассматривать как результат увеличения электрического сопротивления (рис. 8).
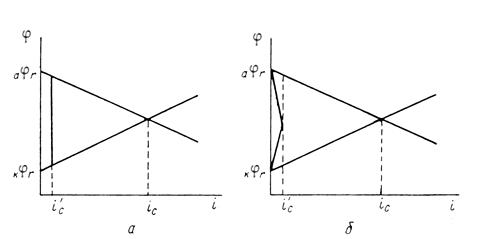
Рис. 3. Коррозионная диаграмма, показывающая, что уменьшение скорости коррозии при нанесении поверхностных защитных слоёв может быть объяснено как увеличением омического сопротивления (а), так и повышением частных коррозионных реакций (б).
Повышение устойчивости железных и стальных изделий при покрытии их поверхности осадками других металлов обусловлено и механической изоляцией поверхности, и изменением ее электрохимических свойств. При этом может наблюдаться или смещение обратимого потенциала анодной реакции в сторону более положительных значений (покрытия медью, никелем, родием), или увеличение поляризации катодной реакции – повышение водородного перенапряжения (цинк, олово, свинец). Как следует из диаграмм (рис.8), все эти изменения уменьшают скорость коррозии.
Обработку поверхности металлов применяют для предохранения машин, оборудования, аппаратов и предметов домашнего обихода при временной защите в условиях транспортировки, хранения и консервации (смазка, пассивирующие пленки) и для более длительной защиты при их эксплуатации (лаки, краски, эмали, металлические покрытия). Общим недостатком этих металлов является то, что при удалении (например, вследствие износа или повреждения) поверхностного слоя скорость коррозии на поврежденном месте резко возрастает, а повторное нанесение защитного покрытия не всегда бывает возможно.
В этом отношении легирование является значительно более эффективным (хотя и более дорогим) методом повышения коррозионной стойкости металлов. Примером повышения коррозийной стойкости металла легированием являются сплавы меди с золотом. Для надежной защиты меди необходимо добавлять к ней значительное количество золота (не менее 52,2 ат.%). Атомы золота механически защищают атомы меди от их взаимодействия с окружающей средой. Несравненно меньше количество легирующих компонентов требуется для повышения устойчивости металла, если эти компоненты способны образовывать с кислородом защитные пассивирующие пленки. Так, введение хрома в количестве нескольких процентов резко увеличивает коррозионную стойкость сталей. Теоретический и практический интерес представляет повышение коррозионной стойкости легированием катодными добавками (Томашов). Для выяснения принципов, на которых основан этот метод, можно, следуя Колотыркину, рассмотреть потенциостатические кривые. В отсутствие внешнего поляризующего тока металл находится при стационарном потенциале (рис. 9), лежащим в области его активного растворения (до легирования). Скорость коррозии определяется при этом пересечением кривых  и соответствует току и соответствует току 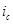 . При введении в исходный металл небольшого количества палладия (или другого металла с низким перенапряжением водорода)поляризационная кривая выделения водорода будет отвечать прямой . При введении в исходный металл небольшого количества палладия (или другого металла с низким перенапряжением водорода)поляризационная кривая выделения водорода будет отвечать прямой 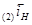 , которая пересечет анодную кривую уже в области пассивного состояния. В результате этого стационарный потенциал сместится в положительную сторону до некоторого значения , которая пересечет анодную кривую уже в области пассивного состояния. В результате этого стационарный потенциал сместится в положительную сторону до некоторого значения 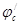 , а скорость коррозии снизится до величины , а скорость коррозии снизится до величины 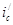 , отвечающей скорости растворения металла в пассивном состоянии. Таким образом, снижение скорости коррозии достигается за счет уменьшения торможений катодного процесса. Такой механизм защиты возможен лишь в том случае, если обратимый потенциал водородного электрода в данных условиях положительнее, чем Фладе – потенциал, и если точка пересечения катодной и анодной поляризационных кривых лежит в области пассивного состояния металла (рис.9). , отвечающей скорости растворения металла в пассивном состоянии. Таким образом, снижение скорости коррозии достигается за счет уменьшения торможений катодного процесса. Такой механизм защиты возможен лишь в том случае, если обратимый потенциал водородного электрода в данных условиях положительнее, чем Фладе – потенциал, и если точка пересечения катодной и анодной поляризационных кривых лежит в области пассивного состояния металла (рис.9).
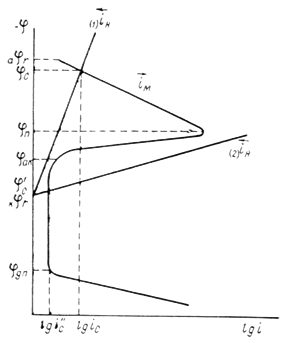
Рис. 4. Поляризационная диаграмма, показывающая возможность защиты пассивирующегося металла от коррозии при увеличении скорости катодного процесса.
Скорость коррозии можно снизить также изменением свойств коррозионной среды. Это достигается или соответствующей обработкой среды, в результате которой уменьшается ее агрессивность, или введением в коррозионную среду небольших добавок специальных веществ, так называемых замедлителей или ингибиторов коррозии.
Обработка среды включает в себя все способы, уменьшающие концентрацию ее компонентов, особенно опасных в коррозионном отношении. Так, например, в нейтральных солевых средах и пресной воде одним из самых агрессивных компонентов является кислород. Его удаляют деаэрацией (кипячение, дистилляция, барботаж инертного газа) или смазывают при помощи соответствующих реагентов (сульфиты, гидразин и т.п.). Уменьшение концентрации кислорода должно почти линейно снижать предельный ток его восстановления, а следовательно, и скорость коррозии металла. Агрессивность среды уменьшается также при ее подщелачивании, снижение общего содержания солей и замене более агрессивных ионов менее агрессивными. При противокоррозионной подготовке воды для уменьшения накипеобразования широко применяется ее очистка ионнообменными смолами.
Ингибиторы коррозии разделяют, в зависимости от условий их применения, на жидкофазные и парофазные или летучие. Жидкофазные ингибиторы делят в свою очередь на ингибиторы коррозии в нейтральных, щелочных и кислых средах. В качестве ингибиторов для нейтральных растворов чаще всего применяются неорганические вещества анионного типа. Их тормозящее действие связано, по-видимому, или с окислением поверхности металла (нитриты, хроматы), или с образованием пленки труднорастворимого соединения между металлом, данным анионом и, возможно, кислородом (фосфаты, гидрофосфаты). Исключение представляют в этом отношении соли бензойной кислоты, ингибирующий эффект которых связан, главным образом, с адсорбционными явлениями. Все ингибиторы для нейтральных сред тормозят преимущественно анодную реакцию, смещая стационарный потенциал в положительную сторону. До настоящего времени еще не удалось найти эффективных ингибиторов коррозии металлов в щелочных растворах. Некоторым тормозящим действием обладают лишь высокомолекулярные соединения.
В качестве ингибиторов кислотной коррозии применяются почти исключительно органические вещества, содержащие азот, серу или кислород в виде амино-, имино-, тиогрупп, а также в виде карбоксильных, карбонильных и некоторых других групп. Согласно наиболее распространенному мнению, действие ингибиторов кислотной коррозии связано с их адсорбцией на границе раздела металл – кислота. В результате адсорбции ингибиторов наблюдается торможение катодного и анодного процессов, снижающие скорость коррозии.
В связи с преобладающим адсорбционным эффектом органических ингибиторов кислотной коррозии особое значение для понимания механизма их действия и для рационального подхода к созданию новых ингибиторов приобретает величина заряда поверхности корродирующего металла, т.е. величина его  - потенциала. Применение приведенной шкалы потенциалов позволяет использовать данные электрокапиллярных измерений на ртути в растворах, содержащих органические соединения, для оченки их эффективности в качестве ингибиторов при кислотной коррозии железа и других металлов. Значение - потенциала корродирующего металла позволяет не только предсказать, какие вещества могут быть ингибиторами, но и рассчитать коэффициенты торможения. Экспериментальные значения коэффициентов торможения кислотной коррозии железа в присутствии различных количеств диэтиламина, сопоставление с расчетной прямой приведены на рис. 10. Расчетная прямая вычерчена по уравнению - потенциала. Применение приведенной шкалы потенциалов позволяет использовать данные электрокапиллярных измерений на ртути в растворах, содержащих органические соединения, для оченки их эффективности в качестве ингибиторов при кислотной коррозии железа и других металлов. Значение - потенциала корродирующего металла позволяет не только предсказать, какие вещества могут быть ингибиторами, но и рассчитать коэффициенты торможения. Экспериментальные значения коэффициентов торможения кислотной коррозии железа в присутствии различных количеств диэтиламина, сопоставление с расчетной прямой приведены на рис. 10. Расчетная прямая вычерчена по уравнению
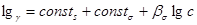 , ,
где  - величина, постоянная для любого члена гомологических рядов аминов и пиридинов, а - величина, постоянная для любого члена гомологических рядов аминов и пиридинов, а 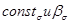 найдены из электрокапиллярных измерений по ртути. найдены из электрокапиллярных измерений по ртути.
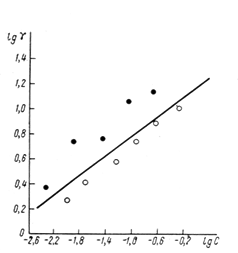
Рис. 5. Сопоставление опытных (--- и --- - данные двух авторов) и расчётных (прямая линия) коэффициентов торможения кислотной коррозии железа при введении разных количеств диэтиланилина.
Адсорбция, однако, является лишь необходимым условием проявления ингибирующего действия органических веществ, но не определяет полностью фактического эффекта ингибиторов. Последний зависит также от многих других факторов – электрохимических особенностей протекания данного коррозионного процесса, характера катодной реакции, величины и природы перенапряжения водорода (при коррозии с водородной деполяризацией), возможных химических превращений ингибитора в ходе коррозии и т.д.
Действие большинства ингибиторов кислотной коррозии усиливается при одновременном введении добавок поверхностно – активных анионов: галогенидов, сульфидов и роданидов.
Парофазные ингибиторы применяются для защиты машин, аппаратов и других металлических изделий во время их эксплуатации в воздушной атмосфере, при транспортировке и хранении. Парофазные ингибиторы вводятся в конвейеры, в упаковочные материалы или помещают в непосредственной близости от работающего агрегата. Благодаря достаточно высокой упругости паров, летучие ингибиторы достигают границы раздела металл – воздух и растворяются в пленке влаги, покрывающей металл. Далее они адсорбируются из раствора на поверхности металла. Тормозящие эффекты в этом случае подобны тем, какие наблюдаются при применение жидкофозных ингибиторов. В качестве парофазных ингибиторов используют обычно амины с небольшим молекулярным весом, в которые введены соответствующие группы, например NО2
или СО2
. В связи с особенностями использования парофазных ингибиторов к ним предъявляются повышенные требования в отношении их токсичности.
Ингибирование – сложный способ защиты, и его успешное применение в различных условиях требует широких познаний.
Глава 3
Обработка резанием.
Обработка резанием является универсальным методом размерной обработки. Метод позволяет обрабатывать поверхности деталей различной формы и размеров с высокой точностью из наиболее используемых конструкционных материалов. Он обладает малой энергоемкостью и высокой производительностью. Вследствие этого обработка резанием является основным, наиболее используемым в промышленности процессом размерной обработки деталей.
1.
Сущность и схемы способов обработки
Обработка резанием — это процесс получения детали требуемой геометрической формы, точности размеров, взаиморасположения и шероховатости поверхностей за счет механического срезания с поверхностей заготовки режущим инструментом материала технологического припуска в виде стружки (рис. 1.1).
Основным режущим элементом любого инструмента является режущий клин (рис. 1.1, а
). Его твердость и прочность должны существенно превосходить твердость и прочность обрабатываемого материала, обеспечивая его режущие свойства. К инструменту прикладывается усилие резания, равное силе сопротивления материала резанию, и сообщается перемещение относительно заготовки со скоростью ν
. Под действием приложенного усилия режущий клин врезается в заготовку и, разрушая обрабатываемый материал, срезает с поверхности заготовки стружку. Стружка образуется в результате интенсивной упругопластической деформации сжатия материала, приводящей к его разрушению у режущей кромки, и сдвигу в зоне действия максимальных касательных напряжений под углом φ. Величина φ зависит от параметров резания и свойств обрабатываемого материала. Она составляет ~30° к направлению движения резца.
Внешний вид стружки характеризует процессы деформирования и разрушения материала, происходящие при резании. Различают четыре возможных типа образующихся стружек: сливная, суставчатая, элементная и стружка надлома (рис. 6).
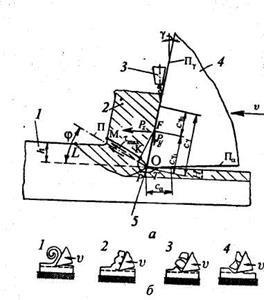
Рис.
6
.
Условная схема процесса резания:
а
– 1
– обрабатываемый материал; 2
– стружка; 3
– подача смазочно-охлаждающих средств; 4
– режущий клин; 5
– режущая кромка; φ – угол сдвига, характеризующий положение условной плоскости сдвига (П) относительно плоскости резания; γ – главный передний угол режущего клина; Р
z
– сила резания; Р
y
– сила нормального давления инструмента на материал; Сγ
u
, Сγ
l
– длины пластичного и упругого контактов; Сγ
, С
a
– длина зон контактного взаимодействия по передней и задней поверхностям инструмента; LOM – область главного упругопластичного деформирования при стружкообразовании; FKPT – область вторичной контактной упруго–пластичнеской деформации металла; h
– глубина резания; Н
– толщина зоны пластического деформирования (наклепа) металла.
В процессе резания режущий клин, испытывая интенсивное трение, контактирует с материалом стружки и обработанной поверхностью в контактных зонах. Для снижения сил трения и нагрева инструмента применяют принудительное охлаждение зоны резания смазочно-охлаждающими средами (СОС), подавая их в зону резания специальными устройствами.
Детали и инструменты закрепляются в специальных органах станка или приспособлениях. Станок, приспособление, инструмент и деталь образуют силовую систему (СПИД), передающую усилие и движение резания от привода станка режущему инструменту и детали.
Реальные схемы различных способов обработки резанием, используемый инструмент, а также виды движения инструмента и заготовки в процессе обработки приведены на рис. 7. В зависимости от используемого типа инструмента способы механической обработки подразделяются на лезвийную и абразивную.
Рис.
7
.
Схемы способов обработки резанием:
а
– точение; б
– сверление; в
– фрезерование; г
– строгание; д
– протягивание; е
– шлифование; ж
– хонингование; з
– суперфиниширование; Dr
– главное движение резания; Ds
– движение подачи; Ro
– обрабатываемая поверхность; R
– поверхность резания; R
оп
– обработанная поверхность; 1 – токарный резец; 2
– сверло; 3
– фреза; 4
– строгальный резец; 5
– протяжка; 6
– абразивный круг; 7
– хон; 8
– бруски; 9
– головка.
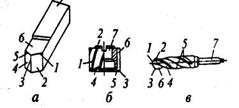
Отличительной особенностью лезвийной обработки является наличие у обрабатываемого инструмента остройрежущейкромки определенной геометрической формы, а для абразивной обработки – наличие различным образом ориентированных режущих зерен абразивного инструмента, каждое из которых представляет собой микроклин.
Рис. 8
Конструкция и элементы лезвийных режущих инструментов:
а
– токарного резца; б
– фрезы; в
– сверла;
1
– главная режущая кромка; 2
– главная задняя поверхность; 3
– вершина лезвия; 4
– вспомогательная задняя поверхность лезвия; 5
– вспомогательная режущая кромка; 6
– передняя поверхность; 7
– крепежная часть инструмента.
Рассмотрим конструкцию лезвийных инструментов, используемых при резании (рис. 8). Инструмент состоит из рабочей части, включающей режущие лезвия, образующие их поверхности, режущие кромки и крепежной части, предназначенной для установки и закрепления в рабочих органах станка.
Основными способами лезвийной обработки являются точение, сверление, фрезерование, строгание и протягивание. К абразивной обработке относятсяпроцессы шлифования, хонингования и суперфиниша. В основу классификации способов механической обработки заложен вид используемого инструмента и кинематика движений. Так, в качестве инструмента при точении используются токарные резцы, при сверлении – сверла, при фрезеровании – фрезы, при строгании – строгальные резцы, при протягивании – протяжки, при шлифовании – шлифовальные круги, при хонинговании – хоны, а при суперфинише – абразивные бруски. Любой способ обработки включает два движения (рис. 1.2.): главное – движене резания Dr
– и вспомогательное – движение подачи Ds
. Главное движение обеспечивает съем металла, а вспомогательное – подачу в зону обработки следующего необработанного участка заготовки. Эти движения осуществляются за счет перемещения заготовки или инструмента. Поэтому при оценках движение инструмента во всех процессах резания удобно рассматривать при неподвижной заготовке как суммарное (рис. 9).
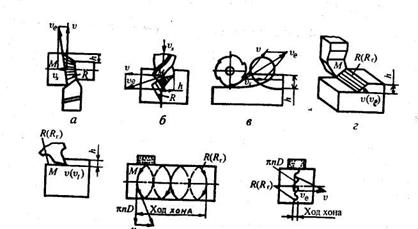
Рис. 9
Схемы определения максимальной скорости режущей кромки инструмента υе
, формы поверхности резания R
и глубины резания h
при обработке:
а
– точением; б
– сверлением; в
– фрезерованием; г
– строганием; д
– протягиванием; е
– хонингованием; ж
– суперфинишированием.
Тогда полная скорость перемещения (ve
)произвольной точки М
режущей кромки складывается из скорости главного движения (v
) и скорости подачи (vs
):
ve
=
v +
vs
(1.1)
Поверхность резания R
представляет собой поверхность, которую описывает режущая кромка или зерно при осуществлении суммарного движения, включающего главное движение и движение подачи. При точении, сверлении, фрезеровании, шлифовании поверхности резания — пространственные линейчатые, при строгании и протягивании — плоские, совпадающие с поверхностями главного движения; при хонин-говании и суперфинишировании они совпадают с поверхностями главного движения.
Поверхности Ro
и Ro
п
называются, соответственно, обрабатываемой поверхностью заготовки и обработанной поверхностью детали
В процессах точения, сверления, фрезерования и шлифования главное движение и движение подачи выполняются одновременно, а в процессах строгания, хонингования движение подачи выполняется после главного движения.
2.
Параметры технологического
процесса резания
К основным параметрам режима резания относятся скорость главного движения резания, скорость подачи и глубина резания.
Скорость главного движения резания (или скорость резания) определяется максимальной линейной скоростью главного движения режущей кромки инструмента. Эта скорость выражается в м/с.
Если главное движение резания вращательное, как при точении, сверлении, фрезеровании и шлифовании, то скорость резания будет определяться линейной скоростью главного движения наиболее удаленной от оси вращения точки режущей кромки — максимальной линейной скоростью главного движения.
v = ω
D/2
(2.1)
где D
-
максимальный диаметр обрабатываемой поверхности заготовки, определяющий положение наиболее удаленной от оси вращения точки режущей кромки, м; ω -
угловая скорость, рад/с.
Выразив угловую скорость ω
через частоту вращения шпинделя станка, получим:
v = π
nD
(2.2)
При строгании и протягивании скорость резания v
определяется скоростью перемещения строгального резца и протяжки в процессе резания относительно заготовки.
При хонинговании и суперфинишировании скорость резания определяется с учетом осевого перемещения (см. рис. 1.4, е, ж
)инструмента.
Скорость резания оказывает наибольшее влияние на производительность процесса, стойкость инструмента и качество обработанной поверхности.
Подача инструмента определяется ее скоростью vs
.
В технологических расчетах параметров режима при точении, сверлении, фрезеровании и шлифовании используется понятие подачи на один оборот заготовки So
и выражается в мм/об. Подача на оборот численно соответствует перемещению инструмента за время одного оборота:
So
= vs
/
n
(2.3)
При строгании подача определяется на ход резца. При шлифовании подача может указываться на ход или двойной ход инструмента. Подача на зуб при фрезеровании определяется числом зубьев Z инструмента и подачей на оборот:
Sz
=
So
/
Z
(2.4)
Глубина резания А определяется расстоянием по нормали от обработанной поверхности заготовки до обрабатываемой, мм. Глубину резания задают на каждый рабочий ход инструмента. При точении цилиндрической поверхности глубину резания определяют как полуразность диаметров до г: после обработки:
h = (
Dur
-
d) / 2
(2.5)
где d
- диаметр обработанной поверхности заготовки, мм. Величина подачи и глубина резания определяют производительность процесса и оказывают большое влияние на качество обрабатываемой поверхности.
К технологическим параметрам процесса относятся геометрия режущего инструмента, силы резания, производительность обработки и стойкость инструмента.
Геометрические параметры режущего инструмента определяются углами, образуемыми пересечением поверхностей лезвия, а также положением поверхностей режущих лезвий относительно обрабатываемой поверхности и направлением главного движения. Указанные параметры идентичны для различных видов инструмента, что позволяет рассмотреть их на примере резца, используемого при точении.
Углы резца по передним и задним поверхностям измеряют в определенных координатных плоскостях. На рис.10изображены координатные плоскости при точении, а на рис. 2.1, б
углы резца в статике.
Главный передний угол γ— угол между передней поверхностью лезвия и плоскостью, перпендикулярной к плоскости резания; главный задний угол α – угол между задней поверхностью лезвия и плоскостью резания; угол заострения β – угол между передней и задней поверхностями. Из принципа построения углов следует, что
α + β + γ = π/2.
Угол наклона режущей кромки X
— угол в плоскости резания между режущей кромкой и основной плоскостью.
Углы в плане: главный угол в плане φ – угол в основной плоскости между следом плоскости резания и направлением продольной подачи; вспомо-
гательный угол в плане φ' – угол в основной плоскости между вспомогательной режущей кромкой и обработанной поверхностью.
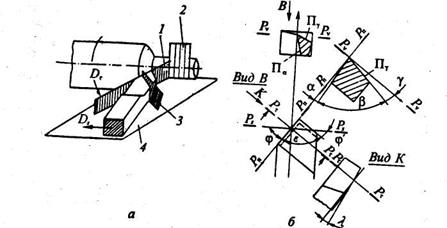
Рис. 10
Геометрические парамеры токарного резца:
а
– координатные плоскости; б
– углы резца в статике;
1
– плоскость резания Рп
; 2
– рабочая плоскость Р
s
; 3
– главная несущая плоскость Р
t
; 4
– основная плоскость Pv
Геометрические параметры режущего инструмента оказывают существенное влияние на усилие резания, качество поверхности и износ инструмента. Так, с увеличением угла у
инструмент легче врезается в материал, снижаются силы резания, улучшается качество поверхности, но повышается износ инструмента. Наличие угла а снижает трение инструмента о поверхность резания, уменьшая его износ, но чрезмерное его увеличение ослабляет режущую кромку, способствуя ее разрушению при ударных нагрузках.
Силы резания Р
представляют собой силы, действующие на режущий инструмент в процессе упругопластической деформации и разрушения срезаемой стружки.
Силы резания приводят к вершине лезвия или к точке режущей кромки и раскладывают по координатным осям прямоугольной системы координат xyz
(рис.11). В этой системе координат ось z
направлена по скорости главного движения и ее положительное направление соответствует направлению действия обрабатываемого материала на инструмент. Ось у
направлена по радиусу окружности главного движения вершины. Ее положительное направление также соответствует направлению действия металла на инструмент. Направление оси х
выбирается из условия образования правой системы координат. Значение усилия резания определяется несколькими факторами. Оно растет с увеличением глубины h
резания и скорости подачи s
(сечения срезаемой стружки), скорости резания ν, снижением переднего угла γ режущего инструмента. Поэтому расчет усилия резания производится по эмпирическим формулам, установленным для каждого способа обработки (см. справочники по обработке резанием). Например, для строгания эта формула имеет вид Р = С
p
hX
p
sY
p
Xn
где коэффициенты Ср
, Хр
,
Yp
,
n
характеризуют материал заготовки, резца и вид обработки.
Мощность процесса резания определяется скалярным произведением:
N
= Pve
(2.6)
Выразив это произведение через проекции по координатным осям, получим:
N
= Pz
vz
+
Py
vy
+ Px
vx
(2.7)
где vx
,
vy
,
vz
— проекции наоси координат скорости движения точки приложения равнодействующей сил резания. В практических расчетах используется приближенная зависимость N =
Pz
v
. Это упрощение обусловлено тем, что составляющие Ру
и Рх
полной силы резания малы по сравнению с Р2
,
а скорость подачи относительно скорости резания составляет всего 1 - 0,1%.
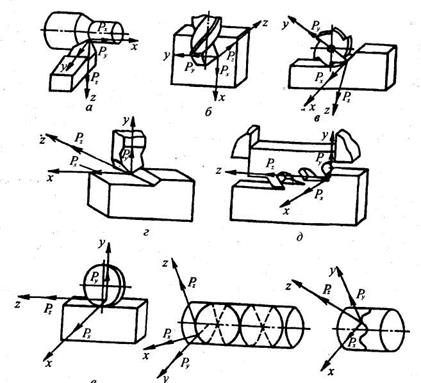
Рис. 11
Схема действия сил резания на режущую кромку инструмента в точке, имеющую максимальную скорость перемещения νе
, при обработке: а
– точением; б
– сверлением; в
– фрезерованием; г
– строганием; д
– протягиванием; е
– хонингованием; ж
– суперфинишированием.
Производительность обработки при резании определяется числом деталей, изготовляемых в единицу времени: Q
= \/Тт
.
Время изготовления одной детали равно Тт
= Тд
+ Тт
+ Ткп
, где То
— машинное времяобработки, затрачиваемое на процесс резания, определяется для каждого технологического способа; Тт
— время подвода и отвода инструмента при обработке одной детали; Гвсп
— вспомогательное время установки и настройки инструмента.
Таким образом, производительность обработки резанием в первую очередь определяется машинным временем То
.
При токарной обработке, мин: То
= La
/(
nso
h
),
где L
- расчетная длина хода резца, мм; а
— величина припуска на обработку, мм.
Отношение a
/
h
характеризует требуемое число проходов инструмента при обработке с глубиной резания И.
Поэтому наибольшая производительность будет при обработке с глубиной резания h
= а,
наибольшей подачей s
0
и максимальной скоростью резания. Однако при увеличениипроизводительности снижается качесто поверхности и повышается износ инструмента. Поэтому при обработке резанием решается задача по установлению максимально допустимой производительности при сохранении требуемого качества поверхности и стойкости инструмента.
Глава 4
Коррозионное растрескивание.
1 Явление коррозийного растрескивания
В металле, подверженном коррозионному растрескиванию, при отсутствии внешних напряжений обычно происходит очень незначительное коррозионное разрушение, а при отсутствии коррозионной среды под воздействием напряжений почти не происходит изменения прочности или пластичности металла. Таким .образом, в процессе коррозионного растрескивания, т. е. при одновременном воздействии статических напряжений и коррозионной среды, наблюдается существенно большее ухудшение механических свойств металла, чем это имело бы место в результате раздельного, но аддитивного действия этих факторов. Коррозионное растрескивание является характерным случаем, когда взаимодействует химическая реакция и механические силы, что приводит к структурному разрушению. Такое разрушение носит хрупкий характер и возникает в обычных пластичных металлах, а также в медных, никелевых сплавах, нержавеющих сталях и др. в присутствии определенной коррозионной среды. При исследовании процесса хрупкого разрушения в результате коррозионного растрескиванир особое значение имеет исследование раздельного воздействия на металл напряжений и коррозионной среды, а также их одновременное воздействие. Однако в процессе коррозионного растрескивания первостепенное значение имеют следующие стадии: 1) зарождение и возникновение трещин и 2) последующее развитие коррозионных трещин. Обе стадии, как будет показано ниже, являются индивидуальными ступенями в процессе коррозионного растрескивания.
2 Коррозионная среда
Средами, в которых происходит коррозионное растрескивание металлов, являются такие среды, в которых процессы коррозии сильно локализованы обычно при отсутствии заметной общей поверхностной коррозии. Интенсивность локализованной коррозии-может быть весьма значительной, в результате чего прогрессирует процесс развития очень узких углублений, достигая, вероятно, наибольшей величины на дне углублений, имеющих радиусы порядка одного междуатомного расстояния. Тщательное изучение литературы показывает, что во многих случаях процесс начальной коррозии может иметь место и при отсутствии напряжений, хотя такое коррозионное разрушение может быть совсем незначительным. Некоторые вопросы, относящиеся к роли напряжений в развитии этих чувствительных зон в определенных системах сплавов, еще остаются неясными, но в общем случае это ясно. Кроме того, большинство экспериментальных работ показывает, что в том случае, когда начальные локализованные коррозионные углубления межкристаллигаы,. то при последующем растрескивании преобладает также межкри-сталлитное разрушение. Если начальная коррозия происходит внутри зерен металла, то последующее растрескивание имеет внутри-кристаллитный характер. Нет определенных указаний о том, что межкристаллитная трещина будет развиваться из внутрикристаллитного коррозионного углубления, и наоборот. Смешанный тип растрескивания, который иногда наблюдается, может быть обусловлен побочным процессом, связанным с динамикой быстро развивающейся трещины.
При воздействии на материал коррозионной среды, которая влияет на склонность сплава к коррозионному растрескиванию и характер разрушения, основными факторами являются следующие:
1) относительная разность потенциалов микроструктурных фаз, присутствующих в сплаве, что вызывает вероятность местного разрушения
2) поляризационные процессы на анодных и катодных участках
3) образование продуктов коррозии, которые оказывают влияние на коррозионный процесс.
3 Структура и состав
Если локализованная коррозия является важным фактором в общем процессе растрескивания, то вполне очевидно, что микроструктура металла должна иметь еще большее значение, определяющее вероятность возникновения такого коррозионного разрушения. Опыты со всей очевидностью показывают, что изменения в составе, термообработке, механической обработке и способах изготовленияприводят к изменению микроструктуры, а, следовательно, влияют и на устойчивость металла против коррозионного растрескивания. Опыты показывают, что структура металла влияет не только на характер начального местного разрушения, но также определяет путь искорость коррозионного растрескивания.
Кроме того, небольшие изменения в составе сплава, без какого-либо очевидного изменения микроструктуры, приводят к заметному изменению устойчивости сплава против коррозионного растрескивания. Например, чистая медь в аммиачных средах не подвержена растрескиванию, но примесь менее чем 0,1 % фосфора, мышьяка или сурьмы в однородном твердом растворе делает ее подверженной разрушению. Добавление 0,3% хрома защищает от коррозионного растрескивания алюминиевый сплав марки 755, что важно в промышленном отношении и что еще раз подчеркивает значение изменений состава сплава на устойчивость против коррозионного растрескивания.
4 Напряжения
Для того чтобы произошел процесс коррозионного растрескивания, необходимо наличие поверхностных или внутренних растягивающих напряжений. Обычно встречающиеся на практике разрушения обусловлены наличием остаточных напряжений, возникающих, при производстве и обработке металла, но в целях исследования не следует делать разграничения между остаточными напряжениями и напряжениями, возникшими в результате приложенных внешних нагрузок. Коррозионное растрескиваниеникогда не наблюдалось в результате действия поверхностных сжимащих напряжений; наоборот, сжиающие поверхностные напряженияразрушения могут использоваться для защиты от коррозионного растрескивания.
При увеличении величины приложенных напряжений уменьшается время до полного разрушения металла. Для коррозионного растрескивания обычно необходимы высокие напряжения, приближающиеся к пределу текучести, однако, часто разрушение может наступить и при напряжениях, значительно меньших предела текучести. Для многих систем сплавов наблюдается какой-то «порог» или «предел» напряжений, т. е. напряжения, ниже которых коррозионное растрескивание не происходит за определенный период времени. Такая зависимость, наблюдавшаяся, например, при замедленном растрескивании сталей, указывает, что основнуюроль в процессе разрушения играют напряжения.
Имеются доказательства, что основное влияние при коррозионном растрескивании напряжения оказывают незадолго до полного разрушения, т. е. эффективность напряжений не сказывается до определенного периода времени, после чего наступает внезапное разрушение. Этот вывод в дальнейшем подчеркивается рядом наблюдений, в которых указывается на зависимость времени до полного разрушения от времени приложения напряжений. Показано, что время до полного разрушения ,не зависит от того, приложены ли напряжения в начале испытания или на последующих стадиях его.
5 Характер коррозионных трещин
Коррозионные трещины развиваются в плоскости, перпендикулярной плоскости растягивающих напряжений, независимо от характера приложенных или остаточных напряжений. С точки зрения микроструктуры коррозионные трещины могут иметь как внутрикристаллитный, так и межкристаллитный характер. Можно предположить, что направление и характер развития трещин в металле до некоторой степени зависят от формы и размера зерен, поскольку эти факторы влияют на распределение внутренних напряжений.
Одно из наиболее важных исследований, относящихся к изучению характера развития трещин, устанавливает, что этот процесс не является непрерывным. На прерывистый характер развития трещин указано в работах Джильберта и Хаддена, Эделеану и Фармери. Обнаружено, что в алюминиево-магкиевых сплавах развитие трещин является ступенчатым процессом, развивающимся путем ряда отдельных механических изломов. Более новое доказательство прерывистого характера развития трещин показано в кинофильме, подготовленном Престом, Беком и Фонтана, занимающимися коррозионным растрескиванием магниевых сплавов.
6 Предотвращение коррозионного растрескивания
Наиболее эффективный метод повышения устойчивости металлов против коррозионного растрескивания состоит в использовании соответствующих конструктивных мероприятий и способов обработки, сокращающих до минимума величину остаточных напряжений. Если остаточные напряжения неизбежны, успешно может быть применена термообработка, снимающая эти напряжения. Если позволяют условия, может быть использована, например, дробеструйная обработка, вызывающая сжимающие поверхностные напряжения, которые впоследствии дают возможность нагружать материал, не вызывая напряженного состояния поверхности. Одним из методов, который получает все большее признание и который связан с электрохимическим фактором процесса растрескивания, является применение катодной защиты.
Одним из интересных методов исследования взаимодействия напряжений и химических факторов является изучение зависимости величины катодного тока, необходимого для защиты, от величины механических напряжений.
Кроме того, ряд исследований показывает, что начавшееся растрескивание может быть остановлено путем применения катодной защиты.
7 Механизм коррозийного растрескивания
Для объяснения характерных особенностей процесса коррозионного растрескивания необходим обобщенный механизм этого явления, который можно было бы применить для всех металлических систем с учетом всех особенностей в каждом индивидуальном случае разрушения.
Первые объяснения механизма коррозионного растрескивания связывались либо с химическим, либо с механическим фактором и были недостаточны, так как не учитывалось совместное химическое и механическое действие. Большой вклад в вопрос понимания механизма коррозионного растрескивания внесен Диксом и соавторами. Их опыты убедительно показали основную электрохимическую природу коррозионного разрушения, а в обобщенном ими механизме коррозионного растрескивания указывается на роль механических факторов в процессе общего разрушения. Согласно этому механизму, процесс коррозионного растрескивания трактуется следующим образом.
Если в металле происходит развитие местного коррозионного разрушения в виде очень узких углублений, то вполне очевидно, что растягивающие напряжения, перпендикулярные к направлению этих углублений, будут способствовать возникновению концентрации напряжений на дне их, причем чем больше углубления и меньше радиус дна углублений, тем больше будет концентрация напряжений. При таком состоянии металла создаются все условия для разрушения его вдоль этих более или менее протяженных локальных коррозионных разрушений, и поэтому при достаточной концентрации напряжений металл может начать разрушаться за счет механического воздействия. В результате механического разрушения будет обнажаться свежая, незащищенная окисной пленкой поверхность металла, которая, будучи более анодной, подвергается интенсивному воздействию коррозионной среды, что приведет к увеличению тока между дном углублений и неповрежденной поверхностью металла, а, следовательно, и к ускорению коррозии. Ускорение коррозионного процесса вызовет дальнейшее механическое разрушение, и, как результат, увеличится скорость развития трещин благодаря совместному действию коррозионной среды и растягивающих напряжений.
Эта общая картина процесса коррозионного растрескивания серьезно не изменилась при последующих исследованиях, и в настоящее время можно дать более детальную оценку механического действия концентратора напряжений и его роли в процессе разрушения.
Существует мнение, что главная функция напряжений состоит в нарушении поверхностных пленок без разрушения металла и что ускоренное развитие и распространение трещин в основном имеет электрохимическую природу. В пленочных теориях коррозионного растрескивания отмечается, что вопрос о том, будет ли иметь место быстрое развитие трещины, зависит от соотношения скоростей образования пленки и увеличения концентрации напряжений. Если образование пленки может остановить коррозию до того, как концентрация напряжений достигнет значительной величины, то быстрое развитие трещин будет предотвращено, но если концентрация напряжений достигнет критического значения до образования пленки, то- произойдет разрушение.
Несмотря на то, что высокие напряжения и деформация могут разрушать поверхностную пленку и тем самым способствовать локализованной ускоренной коррозии, нет достаточных доказательств, что они играют основную роль или что разрушение пленки является главным фактором, приводящим к развитию трещин. Однако возможно, что разрушение поверхностной пленки, если оно имеет место, может играть важную роль в процессе хрупкого разрушения.
Весьма маловероятно, что наблюдаемое в некоторых случаях очень быстрое развитие трещин и последующее разрушение металла может быть причиной протекания коррозионного процесса. Очень быстрое (почти моментальное) растрескивание может быть воспроизведено в лабораторных условиях при соответствующем выборе состава сплава, термообработки и коррозионной среды. Наблюдения за характером развития трещин показывают, что трещины развиваются преимущественно механическим путем. Скорость развития трещин, хрупкий характер разрушения и другие факторы указывают на основную роль напряжений в общем процессе взаимодействия механических и химических факторов, кроме тех случаев, когда происходит разрушение поверхностной пленки, обеспечивающей доступ коррозионной среды. Новые представления о механизме хрупкого разрушения пластичных металлов и исследование влияния поверхностных пленок на ползучесть и пластическую деформацию указывают на основную роль напряжений в процессе развития трещин и хрупкого разрушения. Вполне вероятно, что. в результате совместного действия напряжений и коррозии происходит процесс пластической деформации, что приводит к хрупкому разрушению металла.
Основные характерные черты такого представления о механизме коррозионного растрескивания содержатся в теориях Дикса и соавторов, а также в работах Киттинга. Впоследствии Джильберт и Хадден развили эти представления более подробно для сплавов Аl — 7% Мg, что дало возможность расширить представления о механизме коррозионного растрескивания, пригодного для всех систем сплавов. Полагают, что такой механизм позволяет объяснять многие наблюдаемые явления, ранее трудно со-гласуемые.
Наиболее вероятными процессами, при которых происходит коррозионное растрескивание, являются следующие:
1. Локализованная электрохимическая коррозия вызывает образование небольших узких трещин в виде отдельных углублений, развивающиеся края которых имеют радиусы кривизны порядка атомных размеров. Трещины могут проходить по границам зерен, как, например, в алюминиевых сплавах или латуни, или через-зерна, как, например, в аустенитных нержавеющих сталях или в магниевых сплавах. Количество образующихся трещин может быть различным, но обычно одна трещина развивается в большей степени, чем другие.
2. По мере развития трещины у ее вершины создается концентрация напряжений. Для пластичных сплавов эта концентрация напряжений не превышает максимальной величины, которая приблизительно в 3 раза больше предела текучести. При достаточно высоких напряжениях у вершины трещины происходит местная пластическая деформация, которая предшествует хрупкому разрушению. В настоящее время установлено, что в пластичных металлах хрупкое разрушение не может иметь места без предшествующей пластической деформации. Действительно, именно деформация металла у развивающегося края трещины вызывает хрупкое разрушение за счет действующих у вершины трещины напряжений.
3. В зависимости от формы образца, способа приложения нагрузки, условий испытания и определенного энергетического состояния металла, свойственного процессу развития хрупкого разрушения, трещина может распространиться через весь образец, вызвав-мгновенное разрушение его, или, распространившись на определенное расстояние, развитие ее может прекратиться. Развитие трещины может быть приостановлено при неблагоприятной для процесса растрескивания ориентации границ зерен, при неоднородности кристаллической решетки или при наличии неметаллических включений; развитие ее может остановиться в результате релаксации напряжений при развитии трещины или при определенном энергетическом состоянии, когда производимая работа деформации будет больше, чем увеличение поверхностной энергии, как отмечено у Ирвина и Орована.
4. Развитие трещины за счет механического разрушения обнажает свежую поверхность металла, и коррозионная среда быстро засасывается в трещину под действием капиллярных сил, в результате чего наступает период интенсивной коррозии. Вполне возможно, что эта стадия интенсивной коррозии способствует развитию; трещины, причем коррозия развивается таким образом, что вызывает разветвление трещины. Однако следует считать, что главным фактором в развитии трещины является механическое воздействие а не электрохимические процессы.
5. Ускоренный процесс коррозии, вызванный действием коррозионной среды на не защищенную пленкой поверхность металла, быстро замедляется вследствие поляризации и повторного образования защитной пленки, что связано с изменением концентрации: электролита внутри трещины.
6. После этого опять преобладают условия, медленно развивающаяся локализованная коррозия продолжается до тех пор, пока не возникнет достаточно высокая концентрация, напряжений, которая вызовет деформацию и развитие трещины. Полный цикл процессов повторяется до тех пор, пока не наступит разрушение вследствие развития трещины или уменьшения поперечного сечения напряженного образца.
Вопрос о том, разрушается ли образец сразу после того как образовалась первая трещина или в результате развития нескольких трещин в течение какого-то периода времени, не является существенным в механизме растрескивания и зависит от формы, размеров и толщины образца, а также от величины напряжений и условий испытания.
Таким образом, представленный выше механизм включает две основные стадии процесса коррозионного растрескивания: период; локализованной электрохимической коррозии и последующий период развития трещин. Если разрушение не происходит очень быcтро, процесс растрескивания включает непродолжительный период интенсивной коррозии. Ниже подробно рассматривается каждая из стадий процесса растрескивания, а также факторы, определяющие эти стадии, и экспериментальные данные, подтверждающие изложенные ранее гипотезы.
8 Начальная стадия локализованной коррозии
Состояние поверхности металла, обеспечивающее развитие интенсивной локализованной коррозии, вероятно, подобно тому состоянию, при котором происходит питтинговая коррозия. Локальное коррозионное разрушение происходит обычно при наличии катодных и анодных микроэлементов, которые способствуют концентрации и ускорению электрохимического процесса. Источниками местных анодных участков могут быть: 1) состав и микроструктурные неоднородности сплава, как, например, многофазные сплавы или включения по границам зерен; 2) значительное искажение границ зерен или других субструктурных границ, по которым могут выделяться растворенные атомы; 3) участки границ зерен, возникшие благодаря местной концентрации напряжений; 4) локальное разрушение поверхностной пленки под действием напряжений; 5) участки, возникшие за счет пластической деформации.
9 Системы сплавов, подверженных межкристаллитному растрескиванию
Алюминиево-медные сплавы
. Браун и соавторы показали, что, в результате выделения по границам зерен CuAl2 примыкающие к границам зерен зоны обедняются медью, в результате чего в растворе хлористого натрия между границами зерен и зернами существует разность потенциалов в 200 мв.
Эти обедненные медью зоны, анодны по отношению к выделившейся фазе CuAl2 и по отношению к самим зернам.
Сплавы А1 — 7% М
g
. Джильберт и Хадден показали, что соединение Мg2
А13
, которое выделяется по границам зерен, в нейтральных и кислых растворах хлористого натрия является анодом по отношению к зернам и к обедненным зонам границ зерен. В этих растворах b
-фаза подвержена избирательной коррозии. В водном растворе едкого натра b-фаза катодна по отношению к телу зерен, и в этом случае не происходит ни избирательной коррозии, ни коррозионного растрескивания, а имеет место только общая коррозия. Эделеану предположил, что подверженность интенсивной избирательной коррозии не обусловлена выделяющейся по границам зерен равновесной фазой, а связана с одним из переходных состояний в процессе старения — выделением или адсорбцией растворенных атомов по границам зерен.
Таким образом, в структуре сплавов, упрочняющихся с выделением второй фазы и подверженных межкристаллитному растрескиванию, имеется три участка с различными электрохимическими характеристиками.
1. Зерна твердого раствора.
2. Выделившаяся по границам зерен фаза (или переходное состояние этой фазы или адсорбированных растворенных атомов) .
3. Обедненные каким-либо компонентом участки твердого раствора, примыкающие к границам зерен.
Мягкие стали
. Паркинс показал, что выделяющиеся по границам зерен карбиды вызывают искажение этих границ. В растворах нитратов искаженные границы зерен феррита анодны по отношению к зернам, в результате чего границы служат местом интенсивной межкристаллитной коррозии. Действие напряжений может еще больше исказить эти границы и сделать эту область более анодной.
Медные сплавы в аммиачных средах
. Чистая медь в аммиачных средах не подвержена растрескиванию, но добавление небольших количеств фосфора, мышьяка, сурьмы, цинка, алюминия, кремния или никеля в качестве легирующих элементов, входящих в однородный твердый раствор, вызывает межкристаллитное растрескивание меди. Оказывается, что наблюдаемая разность потенциалов между границами зерен и зернами и местная межкристаллитная коррозия могут быть обусловлены искажением границ зерен в результате различной ориентации смежных зерен . Робертсон, показал, что концентрация легирующего компонента в меди, вызывающая беспорядочную рекристаллизованную структуру, соответствует концентрации, которая делает сплав подверженным кор-озионному растрескиванию. В однородных системах причиной развития местной межкристаллитной коррозии может быть химическая активность границ зерен, которая зависит от искажения границ и действия напряжений и деформации. Выделение или адсорбция по раницам зерен растворенных атомов будет значительно влиять на искажение и активность границ зерен.
В работе Томпсона и Трэси выведено соотношение между концентрацией легирующего компонента, необходимой для получения подверженного коррозионному растрескиванию сплава, и количеством компонента, которое вызывает межкристаллитную коррозию сплава при отсутствии напряжений. Ни в одном случае не наблюдалось коррозионного растрескивания при легировании компонентами, которые не вызывают первоначальной межкристаллитной коррозии. Интересно отметить, что при значительном увеличении концентрации легирующих компонентов алюминия и кремния (но не выходя из области твердых растворов) сопротивление растрескиванию увеличивается и наблюдается смешанный характер растрескивания — и межкристаллитный и внутрикристаллитный, что несомненно связано с изменением активности границ зерен.
10 Системы сплавов, подверженных внутрикристаллитному растрескиванию
Магниевые сплавы.
На основании изучения зависимости между содержанием железа в магниевых сплавах и их устойчивостью против внутрикристаллитного коррозионного растрескивания было высказано предположение, что железо-алюминиевая составляющая, преимущественно выделяющаяся параллельно определенным кристаллографическим плоскостям, в частности плоскости базиса, может быть катодной фазой. Было показано, что разность потенциалов между соединением Fe—А1 и твердым раствором Мg—А1 в солянохроматных растворах составляет 1в.
Наблюдаемое межкристаллитное растрескивание некоторых магниевых, сплавов в дистиллированной воде, в растворах хроматов и фторидов, возможно, обусловлено присутствием незначительных примесей по границам зерен. Как известно, сопротивление магния общей коррозии зависит от наличия некоторых примесей — таких, как железо, медь, никель и кобальт, которые в этом отношении особенно активны.
Аустенитные нержавеющие стали типа 18-8
. Стабильность аустенитной фазы в нержавеющих сталях зависит в основном от содержания в сплаве никеля и азота. Однако, в сталях типа 18-8 в результате холодной обработки или деформации какая-то часть аустенита может превратиться в мартенсит. Было высказано предположение, что пластинки мартенсита являются анодной фазой в процессе местной коррозии. Это предположение подверглось критике на основании того, что некоторые аустенитные нержавеющие стали, которые даже под влиянием значительной холодной обработки не претерпевают мартенситного превращения, подвержены коррозионному растрескиванию. Кроме того, нержавеющая сталь типа 18-8 подвержена коррозионному растрескиванию в атмосфере пара, содержащего хлориды, при температурах, слишком высоких для мартенситных превращений.
Существенным доказательством электрохимического характера локального коррозионного разрушения, т. е. первой стадии процесса коррозионного растрескивания, является возможность предотвращения растрескивания при катодной поляризации и при деаэрации коррозионной среды для некоторых алюминиево-магниевых сплавов. Удаление кислорода из раствора понижает скорость катодного процесса и тем самым препятствует электрохимическому разрушению.
11 Развитие трещин
Существенным подтверждением гипотез механизма коррозионного растрескивания является более подробное изучение характера развития трещин, в частности, микрокиноскопическое исследование процесса развития трещин. Фильм, заснятый Престом, Беком и Фонтана, показывает, что развитию внутрикристаллитной трещины в магниево-алюминиевых сплавах предшествует волна пластической деформации. Как ранее установлено, для возникновения пластической деформации необходимо наличие небольшой трещины, как источника развития хрупкого разрушения. Кроме того, в результате деформации металла у вершины первоначально образовавшейся трещины должно происходить дальнейшее ее углубление и расширение, что наблюдается в действительности. Степень развития трещины в результате деформации определяется, несомненно, пластическими характеристиками материала, и следует ожидать, что при наличии непрерывной хрупкой фазы для развития трещины потребуется небольшая деформация и расширения трещины совсем не произойдет или будет весьма незначительным.
В результате пластической деформации обычно происходит разрушение защитных поверхностных пленок в трещине, что вызывает ускорение коррозионного процесса. Но разрушение пленки может играть и более существенную роль в процессе деформации. Было показано, что защитные пленки на монокристаллах и в некоторых поликристаллических материалах препятствуют протеканию процессов ползучести и деформации. Было высказано предположение, что такие пленки действуют как барьер при передвижении дислокаций и, следовательно, препятствуют деформации. Концентрация дислокаций под поверхностной пленкой вызывает высокую концентрацию напряжений. Когда пленка разрушается или устраняется химическим путем, дислокации стремятся к передвижению по своему первоначальному направлению, вызывая тем самым самопроизвольную деформацию. Если это передвижение происходит в плоскости развития трещины, должно происходить ее углубление и расширение. Деформация, которая происходит у вершины трещины, очень локализована и трудно обнаруживается обычными методами металлографического анализа. Кроме того, cледует учесть, что деформированные участки и поверхности образующихся трещин подвержены интенсивной коррозии, которая может легко уничтожить все признаки существования деформации.
Имеются данные о том, что напряженные и деформированные металлы более активны, чем ненапряженные. Таким образом, участкам с коррозионными трещинами свойственна большая электрохимическая активность, что приводит к ускорению процесса трещинообразования. Не может быть сомнения, что ускоренный коррозионный процесс вызван разрушением поверхностной пленки и наличием напряженных анодных участков, но мало вероятно, что эти факторы способствуют общему процессу развития трещин. Если они оказывают влияние, то только в основном на возникновение небольших местных трещин, необходимых для создания концентрации напряжений и последующего трещинообразования.
Развитие трещины, которая вызывает разрушение металла, не обязательно происходит в том же месте, где появилась первая мелкая трещина. Форма образца и характер деформации могут быть такими, что основная трещина развивается на некотором расстоянии от первоначально появившейся небольшой трещины.
Ирвин и соавторы характеризуют процесс развития трещины следующим образом. Первоначальное зарождение трещин происходит на разрозненных, не связанных между собой участках с большими растягивающими напряжениями. Мелкие разрозненные трещины, соединяясь, образуют одну трещину. Как отмечается, процесс трещинообразования начинается на ослабленных участках металла, и первые стадии его сопровождаются пластической деформацией. Развитие трещины носит прерывистый характер, и это является основным свойством быстрого трещинообразования.
Быстрое начало и прекращение отдельных процессов прерывистого трещинообразования производит на металл сильное механическое воздействие, что может вызвать дальнейшее развитие и разветвление трещины. Развитие трещины приостановится, когда она достигнет области, где нет достаточных растягивающих напряжений, направление которых перпендикулярно направлению развития трещины. Следовательно, для последующего развития трещины необходима дальнейшая деформация. Большие трещины обладают способностью развиваться быстрее, чем мелкие. По мере роста мелких трещин крупные трещины развиваются значительно быстрее, и вскоре начинает преобладать только одна трещина, которая останавливает развитие других. Характер развития трещин в пластичных металлах обеспечивает возникновение ряда быстро развивающихся трещин, так как новые трещины образуются по ходу .продвижения основной трещины и соединяются с ней. Когда энергия деформации, выделяющаяся при развитии основной трещины, становится равной работе деформации, происходит процесс быстрого саморастрескивания. В эту главу не входит подробное обсуждение работ Ирвина и Орована об относительном равновесии между совершённой работой и энергией, освобождающейся при развитии трещины. Следует указать, что если энергия деформации, выделяющаяся при развитии трещины, больше энергии, необходимой для нового разрушения поверхности, трещина будет развиваться самопроизвольно. Ирвин также показал, что скорость развития трещины будет увеличиваться до тех пор, пока не будет достигнуто неустойчивое состояние, после чего произойдет быстрое разрушение.
Изложенное рассмотрение процесса возникновения и развития трещины более точно характеризует природу коррозионного растрескивания. На прерывистый характер развития трещин указываютЭделеану, Джильберт и Хадден, Фармери для алюминиевых сплавов, а также Преет, Бек и Фонтана для магниевых сплавов. Очевидно, нет сомнения, что при растрескивании материал подвергается серии отдельных механических разрушений, которые, соединяясь вместе, образуют трещину. Кроме того, фильмы и микрофотографии, имеющиеся в литературе, показывают, что растрескивание происходитпутем продвижения развивающейся трещины. Можно ожидать, что изложенный механико-электрохимический механизм коррозионного растрескивания может достаточно точно объяснить наблюдаемые явления процесса коррозионного растрескивания, среди которых основными являются следующие:
1. Трещины не возникают и не развиваются под действием сжимающих напряжений.
2. Более высокие напряжения, особенно напряжения, близкие к пределу текучести, вызывают более высокую концентрацию напряжений и соответственно уменьшают устойчивость металла против растрескивания.
3. Для создания достаточной концентрации напряжений и последующей деформации необходим какой-то минимум напряжений, тот минимальный предел напряжений не является абсолютной величиной и зависит от формы образца и условий испытания. Следует также указать, что предел упругости или текучести на отдельных микроскопических участках может быть значительно ниже, чем текучесть сплава.
4. В том случае, когда разрушение металла происходит почти сразу после образования первоначальной трещины, время до растрескивания зависит от времени, необходимого для зарождения мелких коррозионных трещин. Важным фактором является также состояние поверхности. При разрушении, включающем ряд повторных циклов процесса растрескивания, общее время до разрушения определяется как суммарное время образования серии коррозионных трещин. Не наблюдается значительного отличия во времени до разрушения образцов, нагруженных в течение всего испытания, и образцов, нагруженных незадолго до разрушения; время, необходимое для коррозионного растрескивания, не зависит существенно от условий создания напряженного состояния металла.
5. Доказательством того, что наибольшее влияние приложенные напряжения оказывают незадолго до разрушения, служит самопроизвольное растрескивание металла после зарождения первоначальной трещины. Если процесс растрескивания происходит за счет образования серии мелких трещин и по мере развития трещины металл приближается к неустойчивому состоянию, то при наличии деформированных участков металлапроизойдет самопроизвольное развитие трещины и полное разрушение металла.
6. Катодная защита препятствует развитию локальных коррозионных разрушений. При наложении катодного тока увеличиваются радиусы возникающих коррозионных углублений, в результате чего коррозионный процесс может происходить только при увеличении напряжений. Поэтому для предотвращения коррозионного растрескивания при повышенных напряжениях должна применяться более эффективная защита, которая будет препятствовать возникновению локальных коррозионных разрушений и созданию концентраторов напряжений.
Полагают, что если развитие трещины достигнет такого значения, что создаются условия для самопроизвольного растрескивания, то применение катодной защиты не окажет никакого влияния.
7. Если время до растрескивания относительно мало и развивается только одна или несколько трещин, то не наблюдается существенного отличия в коррозии (в количестве металла, переходящего в раствор) напряженных и ненапряженных образцов, как показал, например, Эделеану для сплава А1—7% Мg, так как развитие трещин идет практически только за счет механического разрушения. С другой стороны, процесс химического разрушения приводит к переходу в раствор измеримого количества металла, но переход металла в раствор не будет существенно зависеть от времени до разрушения.
8. Предложенный механизм растрескивания согласуется с наблюдаемым явлением, обнаруживающим одинаковую скорость развития образовавшихся трещин в материале, подверженном коррозионному растрескиванию, и в сравнительно устойчивом материале. Зависимость устойчивости металла против коррозионного растрескивания от его структуры и коррозионной среды в значительно мольшей степени проявляется в первый период зарождения локального разрушения, чем при последующей стадии развития трещин.
9. Чем меньше размер зерна металла, тем больше его устойчивость против коррозионного растрескивания. При увеличении размера зерна уменьшается время до разрушения. Казалось бы, что чем больше число зерен, тем больше число границ зерен, имеющих высокую электрохимическую активность, в результате чего более вероятен процесс локального коррозионного разрушения; однако при мелкозернистой структуре условия для зарождения трещин довольно неблагоприятные. Доказано, что сопротивлениехрупкому разрушению поликристаллических металлов обратно пропорционально квадратному корню размера зерна. Следовательно, для разрушения мелкозернистого поликристаллического материала требуются повышенные напряжения. Поэтому крупнозернистые металлы с благоприятной ориентацией границ зерен очень неустойчивы против коррозионного растрескивания.
В случае межкристаллитного растрескивания большое значение имеет выделение растворенных атомов по границам зерен, так как предполагается, что адсорбция растворенных атомов по границам зерен уменьшает энергию границ зерен и снижает напряжения, необходимые для того, чтобы вызвать хрупкое разрушение (т. е. снижает работу, необходимую для образования новой поверхности). Любой адсорбционный процесс на участках металла с несовершенной структурой, который уменьшает работу, необходимую для образования новой поверхности, значительно увеличивает тенденцию таких участков к трещинообразованию при наличии напряжений.
Очевидно, следует предположить, что хрупкое межкристаллитное растрескивание сплавов вызвано содержанием по границам зерен интерметаллических фаз; в этом случае существуют очень благоприятные условия для развития по границам зерен местной коррозии, а развитие хрупкого разрушения происходит за счет интерметаллической фазы. Для однородных твердых растворов, в которых имеет место межкристаллитное растрескивание (например, в а
-латуни), определяющим фактором является адсорбция или выделение растворенных атомов по границам зерен.
12 Общие закономерности явления коррозийного растрескивания
Вполне очевидно, что сплавы, основу которых составляют благородные металлы, являются наиболее устойчивыми против коррозионного растрескивания, так как легирующие компоненты таких сплавов всегда менее благородий. Кроме того, для таких сплавов ограничено число коррозионных сред, в которых может происходить растрескивание. С другой стороны, для такого очень активного металла, как магний, все легирующие компоненты более благородны, поэтому магниевые сплавы сильно подвержены коррозионному растрескиванию. Для магния даже вода является активной коррозионной средой.
Среди специальных групп сплавов, не подверженных коррозионному растрескиванию, можно отметить сплавы золота, палладия и платины.
Однако для сплавов серебра условия для коррозионного растрескивания более благоприятны. Во-первых, серебро часто используется в виде сплава с более благородными металлами, такими, как золото, палладий и платина; во-вторых, серебро быстро вступает .в реакцию с сильными окислителями, такими, как азотная и хромовая кислоты, а также с соляной кислотой и хлорным железом. Предел устойчивости для этих сред лежит примерно при 40 ат. % Аu, так что коррозионное растрескивание будет иметь место во всех сплавах с золотом, содержащих менее 58,5 вес. % золота. В связис этим имеются многочисленные примеры коррозионного растрескивания сплавов серебра, содержащих золото и палладий, применяющихся в зубоврачебном деле. Коррозионное растрескивание этих сплавов наблюдалось после очистки их в соляной кислоте в процессе производства. С другой стороны, сплавы серебра, содержащие менее благородные компоненты, не подвержены коррозионному растрескиванию. Это подтвердилось при испытании однородных сплавов системы Аu—Zn, содержащих 25 ат. % Zn, и сплавов системы Ag—А1, содержащих 13 ат. % А1. Образцы из этих сплавов, испытываемые под напряжением в 2%-ном растворе FeС3, не подвергались коррозионному растрескиванию даже в течение продолжительного времени испытания.
Медные сплавы более подвержены коррозионному растрескиванию. Число более благородных легирующих компонентов для меди не меньше, чем для серебра, но основная опасность обусловлена тем, что в любой среде, содержащей хотя бы незначительное количество аммиака, происходит коррозионное растрескивание медных сплавов. Все сплавы, содержащие небольшое количество золота, использующиеся в производстве ювелирных изделий, являются сплавами на медной основе. Из всех использующихся сплавов меди с золотом только один сплав, содержащий 75 вес.% золота, не подвержен коррозионному растрескиванию. Для остальных сплавов их устойчивость зависит от коррозионной среды и предела устойчивости.
Предел устойчивости для сплавов системы Сu—Аu в растворе аммиака составляет примерно 20 ат.% Аu, так что сплавы, содержащие 50 вес. % золота, не подвержены коррозионному растрескиванию в этой среде. Но в 2 %-ом растворе FeС3 такие сплавы, содержащие меньше 35 ат.% Аu, подвержены коррозионному растрескиванию. Поэтому в ювелирном деле следует применять сплавы, содержащие не менее 58,5 вес.% золота, с использованием их при возможно более низких напряжениях. Однако в случае технического использования, включающего наличие в сплаве внутренних напряжений и воздействие коррозионной среды (например, изготовление перьев для авторучек), такие сплавы недостаточно устойчивы. Но если часть меди в этих сплавах заменять серебром (по весу), как это часто делается на практике, то атомная доля золота в сплаве увеличивается и соответственно увеличивается сопротивление коррозионному растрескиванию.
Особое внимание следует уделить такому важному сплаву, как латунь, где медь легирована менее благородным компонентом (цинком) и поэтому, согласно правилам, не должна подвергаться коррозионному растрескиванию. Однако практически очень часто происходит сезонное растрескивание латуни. Тщательное исследование показывает, что коррозионное растрескивание латуни происходит только в аммиачных средах, тогда как раствор FeС3, концентрированные кислоты НС1 и НNОз вызывают только общую поверхностную коррозию.
Это отклонение от правил вызвано особым поведением меди в аммиачной среде, в которой образуются комплексные ионы с медью, что вызывает характерный тип коррозии, не свойственной другим средам. В растворах цианистого калия также образуются комплексные ионы, но в отличие от аммиака эта среда не вызывает коррозионного растрескивания сплавов систем Сu—Аu и Сu—Au, хотя она, подобно царской водке, вызывает коррозию золота.
Все остальные технически пригодные металлы менее благородны, чем водород. Из всех технических металлов наименее благородным является магний, в результате чего все легирующие компоненты всегда более благородны, что и обусловливает сильную подверженность магниевых сплавов коррозионному растрескиванию Даже вода, являющаяся для магния активной коррозионной cредой, при наличии внутренних напряжений может вызвать коррозионное растрескивание магниевых сплавов. Поэтому использование таких сплавов в технике в значительной степени ограничено. В результате небольшой растворимости алюминия и цинка в магнии сплавы магния с этими компонентами пересыщены при комнатной температуре, вследствие чего всегда приходится решать вопрос о том, преобладает ли совершенно однородное состояние сплава или в сплаве имеются субмикроскопические выделения второй фазы, вызывающие коррозионное растрескивание. Однако исследования автора, проводимые с магниевыми сплавами, содержащими примерно 1 ат. % А1 и имеющими совершенно однородную структуру, показали, что, несмотря на отсутствие выделений второй фазы, происходит коррозионное растрескивание сплавов.
Более стойкими против коррозионного растрескивания являются алюминиевые сплавы, особенно если они легированы менее благородным элементом — магнием. Согласно общим правилам, коррозионному растрескиванию подвержены только те алюминиевые сплавы, которые содержат цинк и медь, а сплавы системы А1—Mg не растрескиваются даже в пересыщенном состоянии, что неоднократно подтверждалось исследованиями.
|