Dear Sirs.
Thank you for your assistance.
ON THE PROBLEM OF CRYSTAL METALLIC LATTICE IN THE DENSEST PACKINGS OF CHEMICAL ELEMENTS
Yours faithfully, Н.G FILIPENKА
http://home.ural.ru/~filip/
Grodno
Abstract
The literature generally describes a metallic bond as the one formed by means of mutual bonds between atoms' exterior electrons and not possessing the directional properties. However, attempts have been made to explain directional metallic bonds, as a specific crystal metallic lattice.
This paper demonstrates that the metallic bond in the densest packings (volume-centered and face-centered) between the centrally elected atom and its neighbours in general is, probably, effected by 9 (nine) directional bonds, as opposed to the number of neighbours which equals 12 (twelve) (coordination number).
Probably, 3 (three) "foreign" atoms are present in the coordination number 12 stereometrically, and not for the reason of bond. This problem is to be solved experimentally.
Introduction
At present, it is impossible, as a general case, to derive by means of quantum-mechanical calculations the crystalline structure of metal in relation to electronic structure of the atom. However, Hanzhorn and Dellinger indicated a possible relation between the presence of a cubical volume-centered lattice in subgroups of titanium, vanadium, chrome and availability in these metals of valent d-orbitals. It is easy to notice that the four hybrid orbitals are directed along the four physical diagonals of the cube and are well adjusted to binding each atom to its eight neighbours in the cubical volume-centered lattice, the remaining orbitals being directed towards the edge centers of the element cell and, possibly, participating in binding the atom to its six second neighbours /3/p. 99.
Let us try to consider relations between exterior electrons of the atom of a given element and structure of its crystal lattice, accounting for the necessity of directional bonds (chemistry) and availability of combined electrons (physics) responsible for galvanic and magnetic properties.
According to /1/p. 20, the number of Z-electrons in the conductivitiy zone has been obtained by the authors, allegedly, on the basis of metal's valency towards oxygen, hydrogen and is to be subject to doubt, as the experimental data of Hall and the uniform compression modulus are close to the theoretical values only for alkaline metals. The volume-centered lattice, Z=1 casts no doubt. The coordination number equals 8.
The exterior electrons of the final shell or subcoats in metal atoms form conductivity zone. The number of electrons in the conductivity zone effects Hall's constant, uniform compression ratio, etc.
Let us construct the model of metal - element so that external electrons of last layer or sublayers of atomic kernel, left after filling the conduction band, influenced somehow pattern of crystalline structure (for example: for the body-centred lattice - 8 ‘valency’ electrons, and for volume-centered and face-centred lattices - 12 or 9).
ROUGH, QUALITATIVE MEASUREMENT OF NUMBER OF ELECTRONS IN CONDUCTION BAND OF METAL - ELEMENT. EXPLANATION OF FACTORS, INFLUENCING FORMATION OF TYPE OF MONOCRYSTAL MATRIX AND SIGN OF HALL CONSTANT.
(Algorithm of construction of model)
The measurements of the Hall field allow us to determine the sign of charge carriers in the conduction band. One of the remarkable features of the Hall effect is, however, that in some metals the Hall coefficient is positive, and thus carriers in them should, probably, have the charge, opposite to the electron charge /1/. At room temperature this holds true for the following: vanadium, chromium, manganese, iron, cobalt, zinc, circonium, niobium, molybdenum, ruthenium, rhodium, cadmium, cerium, praseodymium, neodymium, ytterbium, hafnium, tantalum, wolfram, rhenium, iridium, thallium, plumbum /2/. Solution to this enigma must be given by complete quantum - mechanical theory of solid body.
Roughly speaking, using the base cases of Born- Karman, let us consider a highly simplified case of one-dimensional conduction band. The first variant: a thin closed tube is completely filled with electrons but one. The diameter of the electron roughly equals the diameter of the tube. With such filling of the area at local movement of the electron an opposite movement of the ‘site’ of the electron, absent in the tube, is observed, i.e. movement of non-negative sighting. The second variant: there is one electron in the tube - movement of only one charge is possible - that of the electron with a negative charge. These two opposite variants show, that the sighting of carriers, determined according to the Hall coefficient, to some extent, must depend on the filling of the conduction band with electrons. Figure 1.
а) б)
Figure 1. Schematic representation of the conduction band of two different metals. (scale is not observed).
a) - the first variant;
b) - the second variant.
The order of electron movement will also be affected by the structure of the conductivity zone, as well as by the temperature, admixtures and defects. Magnetic quasi-particles, magnons, will have an impact on magnetic materials.
Реклама

Since our reasoning is rough, we will further take into account only filling with electrons of the conductivity zone. Let us fill the conductivity zone with electrons in such a way that the external electrons of the atomic kernel affect the formation of a crystal lattice. Let us assume that after filling the conductivity zone, the number of the external electrons on the last shell of the atomic kernel is equal to the number of the neighbouring atoms (the coordination number) (5).
The coordination number for the volume-centered and face-centered densest packings are 12 and 18, whereas those for the body-centered lattice are 8 and 14 (3).
The below table is filled in compliance with the above judgements.
| Element
|
R
H
.
1010
(cubic metres /K)
|
Z
(number)
|
Z kernel
(number)
|
Lattice type
|
| Natrium
|
Na
|
-2,30
|
1
|
8
|
body-centered
|
| Magnesium
|
Mg
|
-0,90
|
1
|
9
|
volume-centered
|
| Aluminium Or
|
Al
|
-0,38
|
2
|
9
|
face-centered
|
| Aluminium
|
Al
|
-0,38
|
1
|
1
2
|
face-centered
|
| Potassium
|
K
|
-4,20
|
1
|
8
|
body-centered
|
| Calcium
|
Ca
|
-1,78
|
1
|
9
|
face-centered
|
| Calciom
|
Ca
|
T=737K
|
2
|
8
|
body-centered
|
| Scandium Or
|
Sc
|
-0,67
|
2
|
9
|
volume-centered
|
| Scandium
|
Sc
|
-0,67
|
1
|
18
|
volume-centered
|
| Titanium
|
Ti
|
-2,40
|
1
|
9
|
volume-centered
|
| Titanium
|
Ti
|
-2,40
|
3
|
9
|
volume-centered
|
| Titanium
|
Ti
|
T=1158K
|
4
|
8
|
body-centered
|
| Vanadium
|
V
|
+0,76
|
5
|
8
|
body-centered
|
| Chromium
|
Cr
|
+3,63
|
6
|
8
|
body-centered
|
| Iron or
|
Fe
|
+8,
00
|
8
|
8
|
body-centered
|
| Iron
|
Fe
|
+8,00
|
2
|
14
|
body-centered
|
| Iron or
|
Fe
|
Т
=1189K
|
7
|
9
|
face-centered
|
| Iron
|
Fe
|
Т=1189K
|
4
|
12
|
face-centered
|
Cobalt or
|
Co
|
+3,60
|
8
|
9
|
volume-centered
|
| Cobalt
|
Co
|
+3
,60
|
5
|
12
|
volume-centered
|
| Nickel
|
Ni
|
-0,60
|
1
|
9
|
face-centered
|
| Copper or
|
Cu
|
-0,52
|
1
|
18
|
face-centered
|
| Copper
|
Cu
|
-0,52
|
2
|
9
|
face-centered
|
| Zink or
|
Zn
|
+0,90
|
2
|
18
|
volume-centered
|
| Zink
|
Zn
|
+0,90
|
3
|
9
|
volume-centered
|
| Rubidium
|
Rb
|
-5,90
|
1
|
8
|
body-centered
|
| Itrium
|
Y
|
-1,2
5
|
2
|
9
|
volume-centered
|
| Zirconium or
|
Zr
|
+0,21
|
3
|
9
|
volume-centered
|
| Zirconium
|
Zr
|
Т=11
35К
|
4
|
8
|
body-centered
|
| Niobium
|
Nb
|
+
0,7
2
|
5
|
8
|
body-centered
|
| Molybde-num
|
Mo
|
+1,91
|
6
|
8
|
body-centered
|
| Ruthenium
|
Ru
|
+22
|
7
|
9
|
volume-centered
|
| Rhodium Or
|
Rh
|
+0,48
|
5
|
12
|
face-centered
|
| Rhodium
|
Rh
|
+0
,48
|
8
|
9
|
face-centered
|
| Palladium
|
Pd
|
-6,80
|
1
|
9
|
face-centered
|
| Silver or
|
Ag
|
-0,90
|
1
|
18
|
face-centered
|
| Silver
|
Ag
|
-0,90
|
2
|
9
|
face-centered
|
| Cadmium or
|
Cd
|
+0,67
|
2
|
18
|
volume-centered
|
| Cadmium
|
Cd
|
+0,67
|
3
|
9
|
volume-centered
|
| Caesium
|
Cs
|
-7
,80
|
1
|
8
|
body-centered
|
| Lanthanum
|
La
|
-0,80
|
2
|
9
|
volume-centered
|
| Cerium or
|
Ce
|
+1,92
|
3
|
9
|
face-centered
|
| Cerium
|
Ce
|
+1,92
|
1
|
9
|
face-centered
|
| Praseodymium or
|
Pr
|
+0,71
|
4
|
9
|
volume-centered
|
| Praseodymium
|
Pr
|
+0,71
|
1
|
9
|
volume-centered
|
| Neodymium or
|
Nd
|
+0,97
|
5
|
9
|
volume-centered
|
| Neodymium
|
Nd
|
+0,97
|
1
|
9
|
volume-centered
|
| Gadolinium or
|
Gd
|
-0,95
|
2
|
9
|
volume-centered
|
| Gadolinium
|
Gd
|
T=1533K
|
3
|
8
|
body-centered
|
| Terbium or
|
Tb
|
-4,30
|
1
|
9
|
volume-centered
Реклама

|
| Terbium
|
Tb
|
Т=
1560К
|
2
|
8
|
body-centered
|
| Dysprosium
|
Dy
|
-2,7
0
|
1
|
9
|
volume-centered
|
| Dysprosium
|
Dy
|
Т=
1657К
|
2
|
8
|
body-centered
|
| Erbium
|
Er
|
-0,341
|
1
|
9
|
volume-centered
|
| Thulium
|
Tu
|
-1,80
|
1
|
9
|
volume-centered
|
| Ytterbium or
|
Yb
|
+3,77
|
3
|
9
|
face-centered
|
| Ytterbium
|
Yb
|
+
3,77
|
1
|
9
|
face-centered
|
| Lutecium
|
Lu
|
-0,535
|
2
|
9
|
volume-centered
|
| Hafnium
|
Hf
|
+0,43
|
3
|
9
|
volume-centered
|
| Hafnium
|
Hf
|
Т=2050К
|
4
|
8
|
body-centered
|
| Tantalum
|
Ta
|
+0,98
|
5
|
8
|
body-centered
|
| Wolfram
|
W
|
+0,856
|
6
|
8
|
body-centered
|
| Rhenium
|
Re
|
+
3,15
|
6
|
9
|
volume-centered
|
| Osmium
|
Os
|
<
0
|
4
|
12
|
volume centered
|
| Iridium
|
Ir
|
+3,18
|
5
|
12
|
face-centered
|
| Platinum
|
Pt
|
-0,194
|
1
|
9
|
face-centered
|
| Gold or
|
Au
|
-0,69
|
1
|
18
|
face-centered
|
| Gold
|
Au
|
-0,69
|
2
|
9
|
face-centered
|
| Thallium or
|
Tl
|
+0,24
|
3
|
18
|
volume-centered
|
| Thallium
|
Tl
|
+0,2
4
|
4
|
9
|
volume-centered
|
| Lead
|
Pb
|
+0,09
|
4
|
18
|
face-centered
|
| Lead
|
Pb
|
+
0,09
|
5
|
9
|
face-centered
|
Where Rh is the Hall’s constant (Hall’s coefficient)
Z is an assumed number of electrons released by one atom to the conductivity zone.
Z kernel is the number of external electrons of the atomic kernel on the last shell.
The lattice type is the type of the metal crystal structure at room temperature and, in some cases, at phase transition temperatures (1).
Conclusions
In spite of the rough reasoning the table shows that the greater number of electrons gives the atom of the element to the conductivity zone, the more positive is the Hall’s constant. On the contrary the Hall’s constant is negative for the elements which have released one or two electrons to the conductivity zone, which doesn’t contradict to the conclusions of Payerls. A relationship is also seen between the conductivity electrons (Z) and valency electrons (Z kernel) stipulating the crystal structure.
The phase transition of the element from one lattice to another can be explained by the transfer of one of the external electrons of the atomic kernel to the metal conductivity zone or its return from the conductivity zone to the external shell of the kernel under the influence of external factors (pressure, temperature).
We tried to unravel the puzzle, but instead we received a new puzzle which provides a good explanation for the physico-chemical properties of the elements. This is the “coordination number” 9 (nine) for the face-centered and volume-centered lattices.
This frequent occurrence of the number 9 in the table suggests that the densest packings have been studied insufficiently.
Using the method of inverse reading from experimental values for the uniform compression towards the theoretical calculations and the formulae of Arkshoft and Mermin (1) to determine the Z value, we can verify its good agreement with the data listed in Table 1.
The metallic bond seems to be due to both socialized electrons and “valency” ones – the electrons of the atomic kernel.
Literature:
1) Solid state physics. N.W. Ashcroft, N.D. Mermin. Cornell University, 1975
2) Characteristics of elements. G.V. Samsonov. Moscow, 1976
3) Grundzuge der Anorganischen Kristallchemie. Von. Dr. Heinz Krebs. Universitat Stuttgart, 1968
4) Physics of metals. Y.G. Dorfman, I.K. Kikoin. Leningrad, 1933
5) What affects crystals characteristics. G.G.Skidelsky. Engineer № 8, 1989
Appendix 1
Metallic Bond in Densest Packing (Volume-centered and face-centered)
It follows from the speculations on the number of direct bonds ( or pseudobonds, since there is a conductivity zone between the neighbouring metal atoms) being equal to nine according to the number of external electrons of the atomic kernel for densest packings that similar to body-centered lattice (eight neighbouring atoms in the first coordination sphere). Volume-centered and face-centered lattices in the first coordination sphere should have nine atoms whereas we actually have 12 ones. But the presence of nine neighbouring atoms, bound to any central atom has indirectly been confirmed by the experimental data of Hall and the uniform compression modulus (and from the experiments on the Gaase van Alfen effect the oscillation number is a multiple of nine.
Consequently, differences from other atoms in the coordination sphere should presumably be sought among three atoms out of 6 atoms located in the hexagon. Fig.1,1. d, e shows coordination spheres in the densest hexagonal and cubic packings.
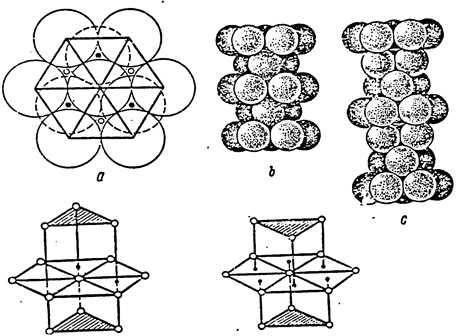
Fig.1.1. Dense Packing.
It should be noted that in the hexagonal packing, the triangles of upper and lower bases are unindirectional, whereas in the hexagonal packing they are not unindirectional.
Literature:
Introduction into physical chemistry and chrystal chemistry of semi-conductors. B.F. Ormont. Moscow, 1968.
Appendix 2
Theoretical calculation of the uniform compression modulus (B).
B = (6,13/(rs
|ao
))5
* 1010
dyne/cm2
Where B is the uniform compression modulus
аo
is the Bohr radius
rs
– the radius of the sphere with the volume being equal to the volume falling at one conductivity electron.
rs
= (3/4 pn ) 1/3
Where n is the density of conductivity electrons.
Table 1. Calculation according to Ashcroft and Mermin
| Element
|
Z
|
rs
/ao
|
theoretical
|
calculated
|
| Cs
|
1
|
5.62
|
1.54
|
1.43
|
| Cu
|
1
|
2.67
|
63.8
|
134.3
|
| Ag
|
1
|
3.02
|
34.5
|
99.9
|
| Al
|
3
|
2.07
|
228
|
76.0
|
Table 2. Calculation according to the models considered in this paper
| Element
|
Z
|
rs
/ao
|
theoretical
|
calculated
|
| Cs
|
1
|
5.62
|
1.54
|
1.43
|
| Cu
|
2
|
2.12
|
202.3
|
134.3
|
| Ag
|
2
|
2.39
|
111.0
|
99.9
|
| Al
|
2
|
2.40
|
108.6
|
76.0
|
Of course, the pressure of free electrons gases alone does not fully determine the compressive strenth of the metal, nevertheless in the second calculation instance the theoretical uniform compression modulus lies closer to the experimental one (approximated the experimental one) this approach (approximation) being one-sided. The second factor the effect of “valency” or external electrons of the atomic kernel, governing the crystal lattice is evidently required to be taken into consideration.
Literature:
Solid state physics. N.W. Ashcroft, N.D. Mermin. Cornell University, 1975
Grodno
March 1996 Н.G. Filipenkа
|