| ОГЛАВЛЕНИЕ
Введение …………………………………………………………………………………………3
1. Определение нуклеотидной последовательности модифицированным методом Максама и Гилберта ……………………………………………………………………………………….4
2. Секвенирование ДНК методом полимеразного копирования ( метод Сэнгера) …………8
3. Филогенетический анализ геномов вирусов ………………………………………………15
4. Компьютерный анализ генетических текстов ……………………………………………..18
Заключение ……………………………………………………………………………………..22
Список литературы …………………………………………………………………………….23
ВВЕДЕНИЕ.
Разработка методов клонирования и определения последовательности оснований (секвенирования) нуклеиновых кислот положила начало новому этапу развития молекулярной биологии. Знание первичной структуры участков генома, выполняющих определенные функции, дало возможность эффективно применить для их исследования целый арсенал новых методов генной инженерии. Эти методы (направленный мутагенез, рекомбинация in vitro и др.) позволяют модифицировать участки нуклеотидных последовательностей и исследовать их функции на молекулярном уровне. С их помощью комбинируются участки генетического материала и создаются геномы с совершенно новыми функциями.
Секвенирование нуклеиновых кислот в настоящее время стало рутинным методом молекулярной биологии. Несомненно, в ближайшем будущем появятся еще более совершенные автоматические секвенаторы, что приведет к резкому увеличению числа расшифрованных последовательностей.
Благодаря знанию генетического кода появилась возможность определять участки нуклеотидных последовательностей, кодирующих потенциальные белки. Этот источник и сегодня дает нам основную информацию о функциональном строении нуклеотидной последовательности.
1. ОПРЕДЕЛЕНИЕ НУКЛЕОТИДНОЙ ПОСЛЕДОВАТЕЛЬНОСТИ МОДИФИЦИРОВАННЫМ МЕТОДОМ МАКСАМА И ГИЛБЕРТА.
Быстрый прогресс, наблюдавшийся в последние годы в различных областях молекулярной биологии, во многом обусловлен появлением эффективного метода определения первичной структуры ДНК. Этот метод, предложенный в 1977 г. Максамом и Гилбертом, основан на селективной химической модификации различных типов гетероциклических оснований в составе ДНК с последующим расщеплением межнуклеотидных связей в модифицированных звеньях. Реакции селективной модификации по каждому типу гетероциклических оснований проводятся таким образом, чтобы в каждой молекуле ДНК в среднем модифицировалось только одно звено данного типа. Поскольку все звенья данного типа в составе молекулы эквивалентны и реагируют с модифицирующим агентом с одинаковыми скоростями, то в сумме каждое звено этого типа окажется частично модифицированным. Дальнейшая обработка ДНК вторичным амином или щелочью приводит к отщеплению модифицированных гетероциклических оснований от цепи ДНК и разрыву полинуклеотидной цепи в местах отщепления гетероциклов (рис. 1).
Реклама

Модификации подвергают ДНК, 32
Р-меченные по 5'-концевому нуклеотидному звену. Радиоактивная метка вводится фосфорилированием с помощью 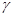 -32
Р-АТР и Т4-полинуклеотидкиназы. Таким образом, в результате химической деградации получается набор фрагментов ДНК различной длины. Длины этих фрагментов соответствуют положению мономерных звеньев того типа, который подвергался модификации. Концевая радиоактивная метка служит точкой отсчета при определении длины продуктов химической деградации ДНК (рис. 2) -32
Р-АТР и Т4-полинуклеотидкиназы. Таким образом, в результате химической деградации получается набор фрагментов ДНК различной длины. Длины этих фрагментов соответствуют положению мономерных звеньев того типа, который подвергался модификации. Концевая радиоактивная метка служит точкой отсчета при определении длины продуктов химической деградации ДНК (рис. 2)
Набор полученных фрагментов фракционируется электрофорезом в ПААГ, который позволяет разделять олиго (поли) нуклеотиды, отличающиеся по длине всего на одно мономерное звено. Последовательность нуклеотидов в ДНК читается непосредственно с радиоавтографа геля.
Метод Максама и Гилберта, разработанный для анализа первичной структуры достаточно длинных ДНК, применим и для коротких (8 – 16 звенных) оли-годезоксирибонуклеотидов. Однако в этом случае реакции химической модификации проводят в более жестких условиях (увеличивая время и температуру реакции) с целью повышения степени модификации.
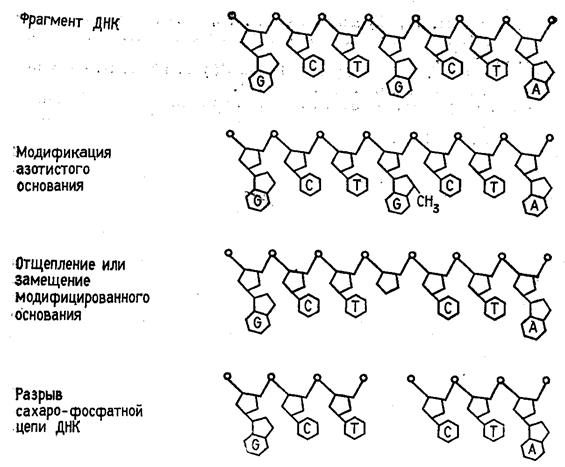
Рисунок 1.1 Отщепление модифицированных звеньев от цепи ДНК после обработки вторичным амином или щелочью.
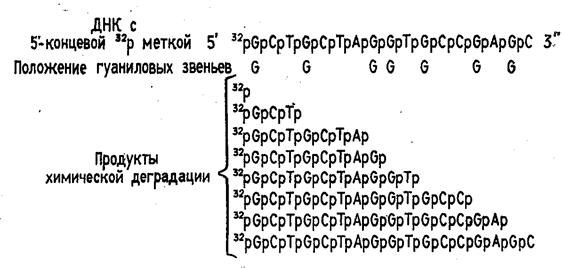
Рисунок 2 Химическая деградация ДНК.
Набор реакций, применяемых для расщепления ДНК по мономерным звеньям определенного типа достаточно велик и постоянно пополняется: по остаткам гуанина – обработка диметилсульфатом (рис. 3); по остаткам аденина и гуанина – апуринизация 50%-ной муравьиной кислотой (по Бартону); по остаткам аденина и цитозина – расщепление гетероциклических оснований под действием 1,2 н. гидроксида натрия и по остаткам тимидина и цитозина – обработка гидразином (рис. 4).
В настоящее время широко используются два основных варианта секвенирования по Максаму — Гилберту. В первом из них реакции химической модификации ДНК проводят в растворе, а во втором ДНК предварительно иммобилизуют на твердой фазе (например, ДЭАЭ-целлюлозе). Первый метод более традиционен, его многочисленные модификации с успехом использовались для секвенирования фрагментов ДНК различных размеров, в том числе олигонуклеотидов. В то же время второй метод имеет ряд преимуществ. Он менее трудоемок и занимает меньше времени, проще в освоении, позволяет обойтись минимальным набором оборудования. В целом оба метода обеспечивают получение вполне приемлемых результатов, а выбор одного из них определяется конкретными условиями лаборатории.
Реклама

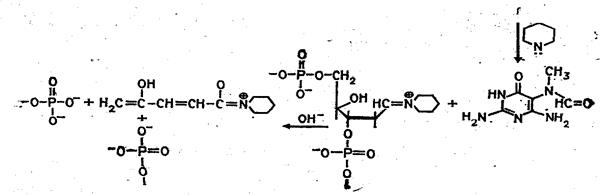
Рисунок 3 Реакция селективного расщепления по остаткам гуанина
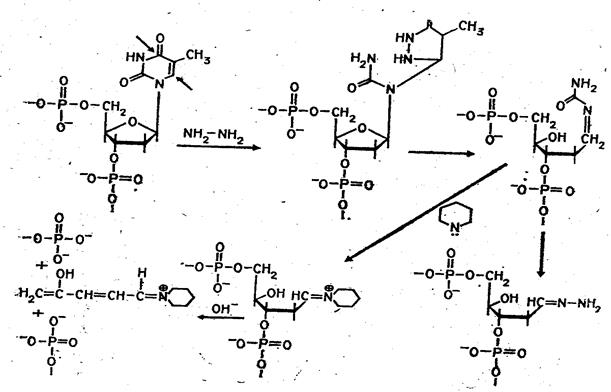
Рисунок 4 Реакция селективного расщепления по остаткам тимидина и цитозина.
2. СЕКВЕНИРОВАНИЕ ДНК МЕТОДОМ ПОЛИМЕРАЗНОГО КОПИРОВАНИЯ.
(МЕТОД СЭНГЕРА)
Ферментативный синтез олиго(поли)дезоксирибонуклеотидов с помощью ДНК-полимераз, заключающийся в копировании матричного полинуклеотида нашел блестящее применение в качестве одного из двух наиболее эффективных методов установления первичной структуры ДНК. Метод состоит в получении блоков-копий полидезоксирибонуклеотида, структура которого изучается. При этом обязательным является выполнение двух условий. Во-первых, копирование должно проводиться, начиная с определенного мономерного звена. Во-вторых, синтез копий следует осуществлять четыре раза, каждый раз останавливая его поочередно на каком-либо одном из четырех мономёрных звеньев (A, G, С или Т), иначе говоря, стремятся получить полный набор "комплементационно отраженных" копий исследуемого полинуклеотида, образование которых прекратилось в каждом из мест расположения одного из четырех мономерных звеньев нуклеиновой кислоты. Определение длины каждой копии позволяет установить положение данного мономерного звена в цепи исследуемого полинуклеотида. Длина копии определяется фракционированием в полиакриламидном геле. Этот метод, таким образом, так же как и метод, основанный на модификации оснований позволяет получать информацию о положении определенного мономерного звена в цепи полинуклеотида прямо после фракционирования.
Для получения копии исследуемого полинуклеотида в последнем выбирают точку отсчета, что достигается введением в систему ферментативного синтеза в качестве нуклеозидного компонента олигонуклеотида-затравки. Такой олигонуклеотид во всех копиях, образующихся в результате достраивания его ферментативным путем, остается постоянным 5'-концевым фрагментом, т. е. является точкой отсчета. Копирование с помощью ДНК-полимеразы в присутствии всех четырех дезоксирибонуклеозид-5'-пирофосфатов (один из них берется  32
Р-меченным) проводят в течение ограниченного времени. Цель этого этапа - проведение статистически ограниченного синтеза для получения всех возможных копий, начиная с затравки, достроенной на одно, два и т. д. звеньев, и включая полную копию изучаемого полинуклеотида. В идеале смесь должна включать все возможные полинуклеотиды (рис. 5), синтез которых статистически прекращается где-то в середине матричного полинуклеотида в районе ATGCTG матричной последовательности. На практике различные компоненты смеси присутствуют в разных количествах. Если такую смесь далее подвергнуть электрофорезу в полиакриламидном геле в определенных условиях, когда скорость движения пропорциональна длине цепи, на электрофореграмме обнаруживается серия полос, представляющих различные олигонуклеотиды. Такое фракционирование обычно не проводят, хотя оно может использоваться для контроля на 32
Р-меченным) проводят в течение ограниченного времени. Цель этого этапа - проведение статистически ограниченного синтеза для получения всех возможных копий, начиная с затравки, достроенной на одно, два и т. д. звеньев, и включая полную копию изучаемого полинуклеотида. В идеале смесь должна включать все возможные полинуклеотиды (рис. 5), синтез которых статистически прекращается где-то в середине матричного полинуклеотида в районе ATGCTG матричной последовательности. На практике различные компоненты смеси присутствуют в разных количествах. Если такую смесь далее подвергнуть электрофорезу в полиакриламидном геле в определенных условиях, когда скорость движения пропорциональна длине цепи, на электрофореграмме обнаруживается серия полос, представляющих различные олигонуклеотиды. Такое фракционирование обычно не проводят, хотя оно может использоваться для контроля на
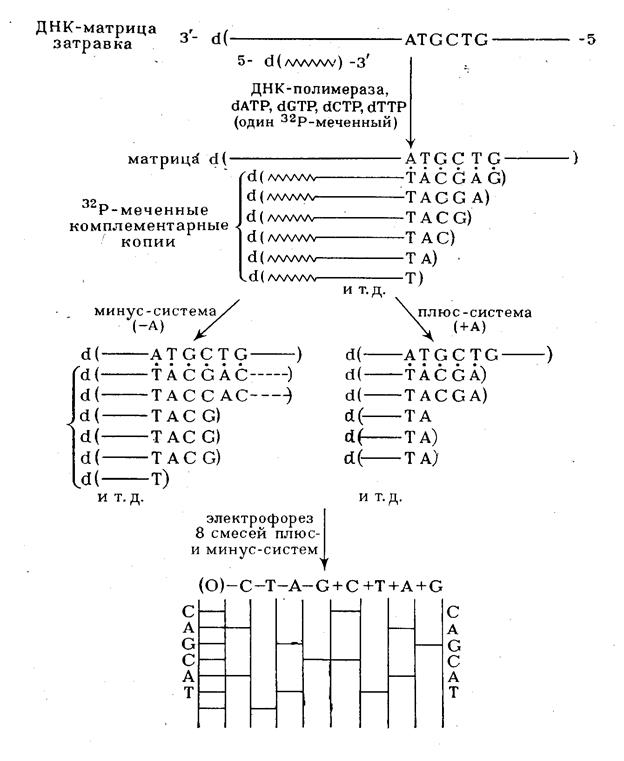
Рисунок 5 Использование ферментативного синтеза олиго(поли)дезоксирибонуклеотидов для определения первичной структуры ДНК.
завершающем этапе анализа. Инкубационную смесь 32
Р-меченных олигонуклеотидов различной длины в виде комплексов с матричным полинуклеотидом подвергают гель-фильтрации для удаления дезоксирибонуклеозид-5'-трифосфатов и аликвоты реакционной смеси реинкубируют с ДНК-полимеразой в различных условиях. В случае "минус-системы" реинкубацию проводят в присутствии только трех дезоксирибонуклеозид-5'-три-фосфатов. Например, в «- А-системе» отсутствует dATP и каждая копия в смеси достраивается с помощью ДНК-полимеразы до места, в котором следующим мономерным звеном должен быть остаток pdA (т.е. Т в матричном полинуклеотиде). Образующуюся смесь фракционируют с помощью электрофореза и обнаруживают (ауторадиографически) ограниченное количество полос (их количество равно количеству Т в матричном полинуклеотиде). Аналогично проводят копирование в отсутствие других субстратов: - dGTP, - dCTP или - dTTP (- G-,- С- и - Т-системы соответственно). Все четыре ионофореза проводят в полиакриламидном геле параллельно. Полученные ауторадиограммы позволяют сразу написать нуклеотидную последовательность, причем чтение цепи снизу вверх соответствует 5'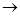 3'-полярности цепи копии. Например, положение самого короткого олигонуклеотида (в " - Т-системе") указывает на то, что следующий за ним по длине олигонуклеотид заканчивается на Т, т. е. что против полосы, расположенной выше (это полоса в - А-системе), следует записать букву Т. Таким же образом записывают далее в последовательности букву А (на основании положения следующего по длине олигонуклеотида, который оказался в "- А-системе") и т. д. Соответствующий участок цепи в матрице читается с учетом принципа комплементарности и антипараллельности цепей в комплексе матрица - затравка. Для проверки этих данных используют результаты анализа с помощью "плюс-системы". В этом случае дополнительное копирование (после первого этапа) проводят в присутствии ДНК-полимеразы, выделенной из бактериофага Т4, которая в отсутствие субстратов (нуклеозид-5'-трифосфатов) проявляет 3'-экзонуклеазную активность (аналогичную действию ФДЭ змеиного яда), т. е. отщепляет мононуклеотиды один за другим с 3'-конца. В то же время в присутствии субстратов ее полимеразная активность во много раз превосходит экзонуклеазную. Так, если в реакционной смеси присутствует хотя бы один дезоксирибонуклеозид-5'-трифосфат (dATP в " +А-системе"), деградация каждой копии, образовавшейся на первой стадии анализа, будет проходить вплоть до места положения А (Т в матрице). В этом случае pdA включается много быстрее, чем удаляется, и, таким образом, накапливаются фрагменты, содержащие на 3'-конце цепи А. Аналогично проводят копирование в присутствии только dGTP, dCTP или dTTP. Смеси параллельно подвергают электрофорезу, как и в предыдущем случае, и получают ауторадиограммы, из которых сразу считывается последовательность 5' 3'-полярности цепи копии. Например, положение самого короткого олигонуклеотида (в " - Т-системе") указывает на то, что следующий за ним по длине олигонуклеотид заканчивается на Т, т. е. что против полосы, расположенной выше (это полоса в - А-системе), следует записать букву Т. Таким же образом записывают далее в последовательности букву А (на основании положения следующего по длине олигонуклеотида, который оказался в "- А-системе") и т. д. Соответствующий участок цепи в матрице читается с учетом принципа комплементарности и антипараллельности цепей в комплексе матрица - затравка. Для проверки этих данных используют результаты анализа с помощью "плюс-системы". В этом случае дополнительное копирование (после первого этапа) проводят в присутствии ДНК-полимеразы, выделенной из бактериофага Т4, которая в отсутствие субстратов (нуклеозид-5'-трифосфатов) проявляет 3'-экзонуклеазную активность (аналогичную действию ФДЭ змеиного яда), т. е. отщепляет мононуклеотиды один за другим с 3'-конца. В то же время в присутствии субстратов ее полимеразная активность во много раз превосходит экзонуклеазную. Так, если в реакционной смеси присутствует хотя бы один дезоксирибонуклеозид-5'-трифосфат (dATP в " +А-системе"), деградация каждой копии, образовавшейся на первой стадии анализа, будет проходить вплоть до места положения А (Т в матрице). В этом случае pdA включается много быстрее, чем удаляется, и, таким образом, накапливаются фрагменты, содержащие на 3'-конце цепи А. Аналогично проводят копирование в присутствии только dGTP, dCTP или dTTP. Смеси параллельно подвергают электрофорезу, как и в предыдущем случае, и получают ауторадиограммы, из которых сразу считывается последовательность 5'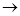 3'-направление, считывается также снизу вверх). Из сравнения фореграмм "плюс-" и "минус-систем" делается однозначный вывод о нуклеотидной последовательности в копиях и, следовательно, в матричном полинуклеотиде. 3'-направление, считывается также снизу вверх). Из сравнения фореграмм "плюс-" и "минус-систем" делается однозначный вывод о нуклеотидной последовательности в копиях и, следовательно, в матричном полинуклеотиде.
В настоящее время выделение фрагментов ДНК, создание рекомбинантных генов, а так же прямое секвенирование ДНК и кДНК становятся общедоступными методами благодаря широкому внедрению ПЦР (полимеразной цепной реакции).Сущность ПЦР заключается в использовании двух олигонуклеотидов-праймеров, способных специфически гибридизоваться с последовательностями нуклеотидов на противоположных концах двух цепей участка ДНК, в качестве затравки для одновременного синтеза комплементарных цепей с противоположных концов матрицы с помощью термостабильной ДНК-полимеразы. В ходе повторяющихся циклов (температурной денатурации ДНК, отжига и энзиматической достройки праймеров) экспоненциально увеличивается количество дискретного фрагмента, фланкированного последовательностями нуклеотидов, соответсвующих первичной структуре праймеров.
Применимость метода Сэнгера зависит от возможности получения одноцепочечных копий клонированных ДНК. Для этой цели можно использовать векторы на основе бактериофага М13. Двухцепочечную чужеродную ДНК можно клонировать в двухцепочечной репликативной форме (РФ) фаговой ДНК, при этом после трансформации в белковую оболочку будет упаковываться только одна из цепей ДНК. Во всех векторах типа М13тр используются сходные полилинкерные последовательности, поэтому для инициации полимеразных реакций пригоден один и тот же универсальный праймер. При амплификации смеси генов (например, семейства генов) необходимо провести клонирование ПЦР-продуктов в векторах типа М13, в результате каждый фаг будет содержать только одну вставку. При прямом секвенировании смеси генов наблюдается несколько одинаково расположенных полос в разных дорожках геля. При амплификации же одного гена можно проводить прямое секвенирование, не прибегая к промежуточному субклонированию.
Выбор оптимального праймера для ПЦР зависит от 5 '- и 3 '-концевых последовательностей амплифицируемого фрагмента ДНК. Кроме того, для встраивания ПЦР-продукта в полилинкерный сайт вектора М13 в 5'-конец праймеров должны быть включены подходящие рестрикционные сайты. В этом случае ПЦР-амплификация с последующей рестрикцией продукта позволит провести его встраивание в ДНК М13, рестрицированную тем же ферментом. В разные концы амплифицируемого фрагмента лучше включать сайты для разных рестриктаз, поскольку это позволит избежать отжига векторной ДНК самой на себя и обеспечит положение клонированной вставки в определенной ориентации (так называемое направленное клонирование). При подборе праймеров необходимо учитывать следующие факторы.
а. Следует убедиться в том, что амплифицируемое семейство генов не содержит консервативного внутреннего рестрикционного сайта, идентичного сайту, включенному в праймер.
б. После включения рестрикционного сайта 5' - конец праймера нужно удлинить, в противном случае рестриктаза не будет расщеплять праймер. Необходимая для каждого фермента длина выступающего участка и время рестрикции указаны в каталоге фирмы New England BioLabs.
Перед секвенированием двухцепочечную рекомбинантную ДНК М13 необходимо перевести в одноцепочечную форму. Для этого ее вводят путем трансформации в компетентные клетки E. с
oli
. Бляшки, содержащие одноцепочечные рекомбинантные фаги, необходимо выколоть, нарастить в бактериальной культуре и депротеинизировать.
Затем переносят культуру в микроцентрифужную пробирку на 1,5 мл и центрифугируют в микроцентрифуге при 12 000 g в течение 5 минут. Переносят 1 мл супернатанта (содержащего чистый фаг) во вторую пробирку на 1,5 мл, добавляют 200 мкл полиэтиленгликоля и инкубируют при комнатной температуре как минимум 15 минут. Собирают фаг центрифугированием в течении 5 минут при 12 000 g и отбирают супернатант. Быстро повторяют центрифугирование и полностью удаляют все следы супернатанта. Затем осаждают ДНК ацетатом натрия, промывают ее 70%-ным этанолом и высушивают под вакуумом. Растворяют ДНК в 30 мкл воды. Полученная ДНК представляет собой одноцепочечную матрицу для секвенирования.
Ниже приведена конкретная методика секвенирования:
Материалы
• 5 х реакционный буфер: 200 мМ трис-HCl, рН 7,5, 100 мМ MgCl2
, 250 мМ NaCl
• Буфер для разведения фермента: 10 мМ трис-HCl, рН 7,5, 5 мМ ДТТ, 0,5 мг/мл БСА
• 5 х смесь для мечения: по 7,5 мкМ dGTP, dCTP, dTTP
• Смесь для ddG-терминации: по 80 мкМ dGTP, dATP, dCTP, dTTP, 8 мкМ ddGTP, 50 мМ NaCl
• Смесь для ddA-терминации: по 80 мкМ dGTP, dATP, dCTP, dTTP, 8 мкМ ddATP, 50 мМ NaCl
• Смесь для ddC-терминации: по 80 мкМ dGTP, dATP, dCTP, dTTP, 8 мкМ ddCTP, 50 мМ NaCl
• Смесь для ddT-терминации: по 80 мкМ dGTP, dATP, dCTP, dTTP, 8 мкМ ddTTP, 50 мМ NaCl
• Стоп-раствор: 90% формамид, 20 мМ ЭДТА, 0,05% бромфеноловый синий, 0,05% ксилолцианол
• Универсальный праймер для секвенирования - 40 (0,5 пмоль/ мкл)
• [35
S]dATP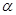 S (1 мКи/37 МБк в 100 мкл) (Amersham, UK; в состав набора не входит) S (1 мКи/37 МБк в 100 мкл) (Amersham, UK; в состав набора не входит)
• 0,1 М ДТТ
Методика
Все реактивы добавляют с помощью диспенсера на 2 мкл Hamilton (PB600), соединенного с адаптером и шприцом 1710 с газовым затвором. Смесь для мечения предварительно разбавляют в пять раз.
1. Для каждой секвенируемой матрицы смешивают в микроцен-трифужной пробирке на 1,5 мл для получения праймерной смеси 6 мкл воды, 1 мкл универсального праймера и 2 мкл реакционного буфера.
2. Размечают микроплашку Falcon 3911. В верхней ее части наносят номера клонов, а слева, сверху вниз, — буквы TCGA.
3. На дно каждой ячейки наносят 2 мкл праймерной смеси, на боковые стенки — по 2 мкл раствора секвенируемой матрицы и центрифугируют плашку. Накрывают ее пленкой Saran® и крышкой и помещают в водяную баню с температурой 70°С на 5 мин. Охлаждают плашку на столе (за это время происходит отжиг праймера и ДНК М13).
4. Пока плашка охлаждается, готовят смесь для мечения. Для этого в микроцентрифужную пробирку на 1,5 мл вносят 0,5 мкл 35
S-dATP, 1 мкл 0,1 М ДТТ, 2 мкл разведенной смеси для мечения и 3,5 мкл воды.
5. Размечают поликарбонатную микроплашку Techne 96® так же, как первую плашку, и в ячейки в ряду "Т" вносят по 2 мкл смеси для ddT-терминации. Аналогичным образом вносят смесь для терминации в ячейки остальных рядов и помещают плашку в термостат для микроплашек с температурой 42°С.
6. После охлаждения плашки (п. 3) в течение 30 мин добавляют к смеси для мечения (для каждой матрицы) последовательно 1,77 мкл буфера для разведения фермента и 0,22 мкл фермента Sequenase® II. (Это позволяет держать фермент Sequenase® II вне холодильника минимальное время.)
7. По 2 мкл этой смеси наносят на боковую стенку ячеек, содержащих праймерную смесь, и центрифугируют плашку для перемешивания компонентов. Включают секундомер.
8. Через 2 мин начинают переносить раствор из ячеек первой плашки в соответствующие ячейки предварительно нагретой и помещенной в термостат поликарбонатной плашки. Для этого используют обычную микропипетку, быстро меняя наконечники после каждой ячейки (помните, что использованные наконечники радиоактивны).
9. После того как перенесен раствор из последней ячейки, включают секундомер и в наконечник на шприце Hamilton набирают стоп-раствор.
10. Через 5 мин наносят по 5 мкл стоп-раствора на боковую стенку каждой ячейки и центрифугируют плашку. После центрифугирования плашку, закрытую крышкой, можно хранить в морозильнике до использования (при - 20°С 35
S-продукты можно хранить в течение недели).
Амплифицированные последовательности нуклеотидов можно увидеть в УФ-свете после фракционирования продуктов ПЦР с помощью гель-электрофореза вприсутствии бромистого этидия. В большинстве случаев после ПЦР при наличии 1 - 10 нг ДНК-матрицы выявляется только одна полоса ДНК ожидаемой электрофоретической подвижности. Чувствительность и специфичность детекции продуктов амплификации значительной увеличиваются при использовании различных вариантов ДНК—ДНК-гибридизации с олигонуклеотидами-зондами, имеющими радиоактивную биотиновую, флюоресцентную или хемолюминесцентную метку. Это сделало возможным проведение работ с минимально возможным количеством материала, (например, с одной клеткой, одной копией гена) без предварительной его очистки.
В качестве исходной матрицы для ПЦР может быть использована ДНК (или кДНК, полученная с помощью предварительной обратной транскрипции РНК), выделенная как из свежеполученных клеток и тканей, так и из замороженных, высушенных или фиксированных препаратов, имеющих частично деградированные нуклеиновые кислоты, т. е. объекты, ранее недоступные для анализа. Так, с помощью методов ПЦР была амплифицирована, клонирована и секвенирована ДНК египетской мумии, продемонстрирована возможность анализа специфических участков ДНК при наличии одного волоса, клетки, сперматозоида в целях идентификации личности и пола хозяина.
Серповидно-клеточная анемия, 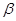 -талассемия, диабет, ревматоидный артрит, мышечная дистрофия, фенилкетонурия, гемофилия, дефицит -антитрипсина - вот далеко не полный список генетических заболеваний, которые могут быть выявлены на ранних стадиях развития эмбриона с помощью ПЦР Разработаны также подходы к раннему выявлению и прогнозированию онкологических заболеваний. -талассемия, диабет, ревматоидный артрит, мышечная дистрофия, фенилкетонурия, гемофилия, дефицит -антитрипсина - вот далеко не полный список генетических заболеваний, которые могут быть выявлены на ранних стадиях развития эмбриона с помощью ПЦР Разработаны также подходы к раннему выявлению и прогнозированию онкологических заболеваний.
3. ФИЛОГЕНЕТИЧЕСКИЙ АНАЛИЗА ГЕНОМОВ ВИРУСОВ.
Филогенетический анализ молекулярных данных является одним из подходов к теоретическому изучению структуры и функции генетических макромолекул (РНК, ДНК, белков) и их эволюционного преобразования. Основная цель филогенетического анализа - изучение эволюционного порядка дивергенции последовательностей генов и белков или их частей, а также восстановление списков эволюционных событий (замен нуклеотидов, делеций и вставок) в предковых линиях этих макромолекул.
Основным инструментом филогенетического анализа является сравнение близких по структуре или по функции генов или белков, и прежде всего, сравнение их первичных последовательностей.
Важнейшим свойством функционально значимых структур макромолекул является их эволюционный консерватизм. Чем меньше функциональная важность отдельных участков генов, тем больше они имеют тенденцию к эволюционной изменчивости. Так, например, псевдогены по-видимому полностью утратили функциональную активность. Для них характерно быстрое накопление в ходе эволюции различных замен, делеций и вставок, разрушающих исходную структуру гена. С другой стороны, гистоны Н4, играющие важную роль в упаковке хроматина, почти не изменялись на протяжении всей эволюции животных.
Консервативность генов позволяет выявить отдаленное родство между их представителями, давно разошедшимися в ходе эволюции и выполняющими иногда разные функции. Однако для филогенетического анализа необходимо и наличие определенного уровня изменчивости генов. Мутации, делеции и вставки являются своего рода метками, благодаря которым удается восстановить пути эволюции современных форм макромолекул. Гены с разной величиной консервативности пригодны для изучения разных эволюционных уровней. Сильно консервативные гены и их продукты (гистоны, тРНК) нельзя , например, использовать для исследования эволюции отрядов и более мелких таксонов, но с успехом можно применять для изучения эволюции более крупных таксонов. Сильно вариабельные гены, наоборот, дают хорошее разрешение лишь на поздних эволюционных этапах.
В последнее время метод полимеразной цепной реакции (ПЦР) с последующим анализом нуклеотидной последовательности широко используется для точной идентификации вирусов и определения их родства в отношении других штаммов. Для сравнительной характеристики геномов различных штаммов вирусов также проводят рестрикционный анализ ПЦР-продуктов. Обычно для построения филогенетического дерева используются данные последовательностей нуклеиновых кислот. Филогенетическое дерево очень ясно показывает родство между вирусами, если анализируется большое количество изолятов. Для этих целей существует много компьютерных программ. Наиболее популярные пакеты программ- PHYLIP(PHYLogeny Inference Package), PAUP(Phylogentic Analysis Using Parsimong),CLUSTAL и MEGA.
Для филогенетического анализа особенно интересны РНК-содержащие вирусы, которые существуют как гетерогенные популяции. Их геном более генетически пластичен, чем геном ДНК-содержащих вирусов.
Так, геном вируса бешенства, который относится к роду Lyssavirus семейства Rhabdoviridae, представлен одноцепочечной негативной РНК длиной около 12000 пар оснований (п.о) , кодирующей пять основных белков.
Белки подразделяются на три функциональные группы: оболочечные (G ,M) нуклеокапсидный (N) и РНК-полимеразный комплекс, состоящий из L и NS белков. Белки N,NS и L вместе с вирионной РНК образуют нуклеокапсид , который окружен мембраной, содержащей трансмембранный гликопротеин G , ответственный за антигенные свойства вируса. Существование псевдогена y между G и L цистронами , является отличительной особенностью вируса бешенства от вируса везикулярного стоматита.
N-ген лиссавирусов, как показало клонирование и секвенирование, является наиболее консервативным по своей структуре из всех генов вируса бешенства.
Mannen K.et al, в 1991 г. определили большую зависимость различий в N-гене от географической локализации, чем от хозяйской специфичности. В Онтарио вирус бешенства, который описан как единственный «Арктический» тип, разделили на четыре основных типа. Эти типы филогенетически разветвляются на две основные ветви , одна из которых состовляет один тип, вторая три основных типа, что отражает историческое передвижение вируса в регионе от середины к концу 50-х гг. Эпизоотия передвигалась на юг Онтарио с севера и Квебека. Изменения в последовательности N-гена , определенных для 4-х вирусных типов, могут представлять генетический маркер для более существенных изменений в других частях вирусного генома.
Kissi et al представили первое сравнение между генотипами и молекулярными различиями N-гена внутри первого генотипа. Филогенетический анализ гена нуклеопротеина 82-х лиссавирусов подтвердил существование шести генотипов лиссавирусов, и также выделил изоляты бешенства первого генотипа в отдельные генетические линии. Изоляты с меньшей чем 80% нуклеотидной и 92% аминокислотной гомологией относятся к различным генотипам. Два коротких региона из 400 нуклеотидов, кодирующих аминоконец N-протеина, и 93 нуклеотида, кодирующих N-NS-регион, могут быть использованы для определения географического распределения основных вирусных линий. Выявлено два региона, имеющих наименьший уровень гомологии: участок длиной 199 пар оснований (нуклеотиды от 1080-го до 1278-го) и более протяженный фрагмент, расположенный между 99-м и 405-м нуклеотидами. Филогенетическое дерево построили, используя пакет программ MEGA. С их помощью получили дерево с ветвями, делящимися на 6 кластеров, которые соответствуют 6-ти генотипам. Сравнение изолятов показало, что в генетическом плане наиболее близкими оказались 4-й и 5-й генотипы, у которых уровень различий составил 79,8% (нуклеотидный уровень) и 93,3% (аминокислотный уровень) [Duvenhage virus (4) и EBL1(5)]. Внутри генотипа 1 наименьшее сходство среди изолятов из Азии и Латинской Америки – 83,3%; и наибольшее сходство в изолятах из Африки и Латинской Америки – 92,2%.
Филогенетический анализ изолятов 1-го генотипа вируса бешенства.
Kissi et al. идентифицировали 11 филогенетических линий, взятых в соответствии с их географическим происхождением и видом хозяина: Африка 1а, Африка 1в, Африка 2, Африка 3, Азия, Арктика, Европа/Средний Восток, Латинская Америка 1 и 2 и две группы вакцинных штаммов. Филогенетическое дерево строилось на основании сравнения фрагмента или целого N-гена.
Этот анализ позволил более точно определить циркуляцию по зонам и установить происхождение и распространение бешенства для некоторых линий, которые, возможно, произошли независимо на этом континенте от различных предшественников. Хотя циркуляция африканских вирусов 1а и 1в более отлично от вирусов, распространенных в Европе и Среднем Востоке, однако была выявлена генетическая связь между ними, что свидетельствует об общем предке.
Выяснено, что накопление большинства нейтральных мутаций в географически разделенных вирусных популяциях привело к значительным расхождениям в нуклеотидной последовательности гена нуклеопротеина.
При исследовании гена нуклеопротеина 11-ти вирусов бешенства японскими учеными было выявлено 9 отдельных кластеров по гомологии менее 90% региона N-гена. Тем самым они подтвердили данные филогенетического анализа, полученные Kissi et al.
Таким образом, варианты вируса бешенства, сгруппированные в соответствии с их географическим распределением, могут быть использованы для исследования эволюционного развития вируса бешенства.
Принципы и методы ОТ-ПЦР.
Обратно-транскриптазная ПЦР состоит из двух этапов:
1- синтез комплементарной ДНК с помощью фермента обратной транскриптазы и затравки
2- амплификация гена или его фрагментов при помощи фермента термостабильной ДНК-полимеразы и коротких олигонуклеотидных 20-30-членных затравок (праймеров), комплементарных 3'- концевым последовательностям антипаралельных цепей ДНК гена. Повторяя стадии денатурации, отжига праймеров и полимеризации (достройка праймеров) 30-35 раз за 2-3 часа получают миллионы копий специфического участка генома вирусов и бактерий.
Анализ ПЦР-продуктов.
Аликвоты ПЦР-продуктов разрезают соответствующими ферментами и разделяют в 1%-м агарозном геле. Учет результатов проводят по размеру ПЦР-продуктов с помощью электрофореза в агарозном геле. На основании размеров и расстояний пробегов маркерных ДНК вычисляют размеры исследуемых фрагментов ДНК.
4. КОМПЬЮТЕРНЫЙ АНАЛИЗ ГЕНЕТИЧЕСКИХ ТЕКСТОВ.
Выявление и анализ закодированных в последовательностях функциональных сигналов требует применения современных методов информатики - качественных баз данных с современными средствами управления, новейших методов распознавания образов, статистических исследований, применения специальных алгоритмов для преодоления возникающих вычислительных трудностей.
В настоящее время исследование функциональных свойств расшифрованных последовательностей нуклеиновых кислот - это новый раздел молекулярной биологии, граничащий с информатикой, с одной стороны, и молекулярной биофизикой - с другой. Можно с уверенностью сказать, что в настоящее время анализ последовательности биополимера позволяет извлечь лишь очень небольшую долю закодированной в ней информации. В конечном счете точное выявление функциональных особенностей в последовательностях нуклеиновых кислот будет возможно только после детального исследования соответствующих реакций, осуществляемых нуклеиновобелковыми комплексами.
Для оперативной работы с последовательностями создаются специальные банки данных. В банке в доступном для пользователя виде хранится каждая расшифрованная последовательность и ее паспорт, в котором указаны различные сведения о ней. Это сведения об организме, из которого выделена последовательность, о документе, где она описана, о расположении на ней регуляторных участков и белках, которые она кодирует и т.д. В настоящее время созданы три большие базы данных последовательностей нуклеиновых кислот: "Genbank" (Лос-Аламос, США - более 30 млн. нуклеотидов), база данных нуклеотидных последовательностей Европейской молекулярно-биологической лаборатории (EMBL, Гейдельберг, ФРГ - более 30 млн. нуклеотидов) и "Генэкспресс" (СССР, ВИНИТИ-ИМГ АН СССР - более 11 млн. нуклеотидов). Известны также несколько белковых баз данных, наиболее представительной из которой является MBRF-PIR (США). Эти базы данных распространяются на различных носителях - магнитных лентах и дисках, на оптических дисках.
Кроме построения филогенетических древ геномов вирусов компьютерный анализ применяется при поиске гомологий, распознавании кодирующих областей, функциональных сигналов, физическом (рестрикционном) картировании молекул ДНК и для предсказания вторичных структур РНК.
Сейчас в мире создано большое количество программ ( обычно организованных в пакеты ) , предназначенных для анализа последовательностей нуклеиновых кислот и избавляющих исследователей от многих трудоёмких рутинных операций , в том числе: подсчёт числа моно -, ди – и тринуклеотидов, перевод нуклеотидной последовательности в аминокислотную и т.д.
Все программы условно делятся на два класса: общего назначения и специального. Первые осуществляют ряд_ наиболее распространенных операций по сбору и анализу последовательностей и позволяют: вводить и редактировать новые последовательности, считывать с помощью сканирующих устройств информацию непосредственно с автографов или гелей', находить участки узнавания эндонуклеаз рестрикции и представлять результаты в удобном (табличном или графическом) виде, находить участки с элементами поворотной и зеркальной симметрии (палиндромы), транслировать нуклеотидную последовательность в белковую во всех трех рамках считывания, сравнивать две последовательности методом точечных матриц гомологии, сравнивать новую последовательность со всеми данными Ген Банка, находить участки, обогащенные теми или иными нуклеотидами, вычислять гипотетическую температуру плавления ДНК, осуществлять автоматическую сборку секвенированных фрагментов в единую структуру - молекулу ДНК, транслировать белковую последовательность в нуклеотидную с учетом неравномерности использования кодонов-синонимов, определять молекулярную массу НК и белков, предсказывать вторичную структуру белков, вычислять свободную энергию образования шпилек и др.
Программы специального назначения создаются для решения более специальных и часто более сложных задач и представляют интерес для более узкого круга специалистов. Так, например, они могут выполнять ряд функций: вычисление длины фрагментов ДНК на основании их электрофоретической подвижности в гелях; выбор гибридизационных зондов; предсказание вторичной структуры РНК; локализация нуклеотидов в гене, которые могут быть изменены (без изменения аминокислотной последовательности) с целью введения сайта узнавания эндонуклеазы рестрикции; нахождение участков с потенциально возможной структурой Z-формы ДНК; выявление функционально значимых участков в неизвестной вновь расшифрованной структуре на основании ранее выведенного консенсуса (в результате сравнительного анализа ряда известных структур с одинаковой функцией); локализацию участков, кодирующих белки, и т.д.
Для примера представлено меню пакета программ MICROGENIE, из которых следует, какие функции общего или специального назначения может выбрать исследователь при работе с нуклеотидными последовательностями.
Программа общего назначения "COMMON" обеспечивает ввод последовательности в ЭВМ, а также проверку введенных данных в диалоговом режиме. Они позволяют также редактировать нуклеотидные последовательности: вводить замены и вставки, исключать нуклеотиды, вырезать, встраивать и объединять нуклеотидные последовательности и таким образом моделировать гибридные и мутантные молекулы ДНК. Ввод последовательностей в память машины можно осуществлять вручную с клавиатуры, но в последнее время созданы приборы для автоматического сканирования авторадиограмм и секвениру-ющих гелей, полученных при использовании флуоресцентных меток , передачи данных сразу в компьютер и последующего анализа последовательности с помощью специальных программ. В недавно вышедшей в издательстве IRL (Оксфорд) книге "Анализ сиквенса нуклеотидных кислот и белков" подробно описываются как конструкция сканирующих устройств, так и программы для чтения авторадиограмм. Созданы программы для восстановления первичных структур высокомолекулярных ДНК на базе данных сиквенса фрагментов, полученных при ее Неспецифическом расщеплении (например, ультразвуком,). Родство между любой парой фрагментов ДНК выявляется на основании совпадения последовательности нуклеотидов в их структурах, причем эти совпадающие последовательности и являются местом перекрывания и такие два фрагмента могут быть объединены в более протяженную структуру. Процесс отбора фрагментов и стыковки продолжается до. тех пор, пока не будет восстановлена вся первичная структура исследуемой ДНК. Одной из такого рода программ является "CONTIG" (существует ее вариант для компьютера IBM PC), созданная в лаборатории Ф.Сангера (Кембридж, Англия). Ниже приводятся основные операции, которые позволяет осуществлять программа "CONTIG":
1) хранение сиквенса каждого фрагмента;
2) отбор смежных фрагментов и сборка последовательностей из них;
3) сравнение данных, полученных при чтении новых авторадиограмм, с уже установленными последовательностями;
4) объединение двух фрагментов с помощью третьего, представляющего собой область перекрывания первых;
5) поиск участков ДНК, комплементарных уже установленным, что является проверкой правильности сборки полной структуры ДНК.
ЗАКЛЮЧЕНИЕ.
Метод Максама-Гилберта и метод Сэнгера основаны на одном принципе. В первом используется специфическое расщепление ДНК, обусловленное природой оснований, во втором - статистический синтез ДНК, заканчивающийся на каком-либо одном из 4 нуклеотидов. Таким образом, основой обоих методов является получение полного (статистического) набора фрагментов ДНК, оканчивающихся на каждом из четырёх нуклеотидов.
Химический метод (метод Максама-Гилберта) проще использовать в том случае, когда исследуемая ДНК не слишком велика (200-500 звеньев). В том случае, если речь идет о секвенировании высокомолекулярной ДНК, лучше применять метод полимеразного копирования (метод Сэнгера) , чтобы не вводить процедуру рестриктазного расщепления с выделением индивидуальных фрагментов. При энзиматическом секвенировании протяженных одноцепочечных ДНК (например, бактериофагов) можно применять набор олигонуклеотидов-затравок, синтез которых в настоящее время не требует больших затрат времени и труда. Для двутяжевых высокополимерных ДНК наиболее удобен метод слепого энзиматического секвенирования с применением универсальной затравки (их выпускают многие фирмы) и обработки данных с помощью ЭВМ. Химический метод также может быть применен, но в этом случае необходимо вырезать из вектора исследуемые фрагменты ДНК, и это усложняет всю процедуру.
СПИСОК ЛИТЕРАТУРЫ.
1. Жарких А.А. Методы филогенетического анализа генов и белков //Молек.биология. (Итоги науки и техники. ВИНИТИ; М. 1985)
2. Компьютерный анализ генетических текстов / А.А.Александров, Н.Н.Александров, М.Ю.Бородовский И ДР. М.: Наука.
3. Методы молекулярной генетики и генной инженерии. Отв. ред. Р.И.Салганник.-Новосибирск: Наука. Сиб. отд-ние, 1990.
4. Молекулярная клиническая диагностика.Методы: Пер. С англ. / Под ред. С.Херрингтона, Дж.Макги. – М.: Мир, 1999.
5. Шабарова З.А., Богданов А.А. Химия нуклеиновых кислот и их компанентов. – М.: Химия, 1978.
6. Шабарова З.А., Богданов А.А., Золотухин А.С. Химические основы генной инженерии: Учебное пособие . – М.: Изд-во МГУ, 1994.
7. Экспериментальные методы исследования белков и нуклеиновых кислот / Подред. М.А.Прокофьева. – М.: Изд-во МГУ, 1985.
|
