Реферат по органической химии на тему:
«Промышленный синтез фенола»
Выполнил:
Проверил:
Содержание
Введение
1. Промышленные способы получения фенола
2. Поиск новых путей синтеза фенола
3. Открытие цеолитных катализаторов для окисления бензола закисью азота
4. Природа каталитической активности цеолитов
5. Специфика действия N2
O как окислителя
6. Стехиометрическая реакция бензола с a-кислородом
7. Биомиметические свойства a-кислорода
8. Активное состояние железа в цеолитной матрице
9. Новый фенольный процесс. Технологические аспекты
Заключение
Список литературы
Введение
История фенола насчитывает уже более 160 лет. Впервые он был выделен из каменноугольной смолы в 1834 г., откуда и получил свое название. карболовая (угольная) кислота. Быстрый рост потребления фенола поставил вопрос об искусственных способах его получения, создание которых является одной из самых ярких страниц в истории органической химии. Ниже кратко описаны промышленные процессы, которые в то или иное время использовались для получения фенола.
1. Промышленные способы получения фенола
1. Сульфонатный процесс
был первым фенольным процессом, реализованным в промышленном масштабе фирмой «BASF» в 1899 г.[1]. Этот метод основан на сульфировании бензола серной кислотой с последующим щелочным плавлением сульфокислоты при 300–350 °С:
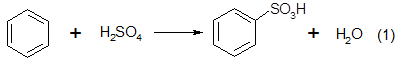
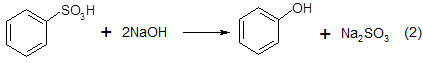
Для сульфирования бензола при получении бензолсульфокислоты применяют концентрированную серную кислоту; соотношение Н2
SО4
: бензол составляет 2:1. Периодический процесс сульфирования проводят при 120—150° С [95].
При щелочном плавлении сульфокислот замещение сульфогруппы на оксигруппу считают результатом сложного процесса, предполагающего промежуточное присоединение щелочи по кратной связи:
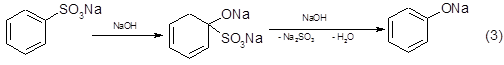
Кроме этого основного процесса возможно также прохождение и других, ведущих к образованию побочных продуктов, в частности протекает расщепление сульфонов с образованием фенолятов и частично дифенола:

Возможно взаимодействие сульфокислот и фенолятов с образованием дифениловых эфиров:
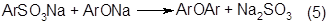
Наконец, возможно окисление компонентов сплава под действием кислорода воздуха с образованием диоксидифенилов, оксидифенилов и их гомологов [2].
Синтез фенолов слагается из ряда стадий, общая схема представлена на рис.1.
Реклама


Рис.1. Схема получения фенолов щелочным плавлением сульфокислот.
Выход фенола в данном процессе зависит от температуры и времени щелочного плавления. Так на 1 тфенола в среднем расходуется несколько более 0,9 т бензола, 1,55 т серной кислоты (в пересчете на 100%-ную), и 1,54 г каустической соды [3].
Несмотря на применение агрессивных реагентов и образование большого количества отходов сульфита натрия, данный метод использовался в течение почти 80 лет. В США это производство было закрыто лишь в 1978 году [95].
2. В 1924 г. фирмой «Dow Chemical» (США) был разработан процесс получения фенола, включающий реакцию хлорирования бензола и последующий гидролиз монохлорбензола
:
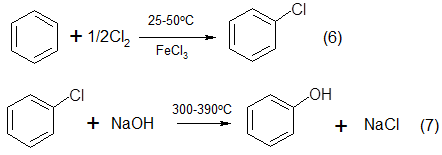
Независимо аналогичная технология была разработана немецкой фирмой «I.G. Farbenindustrie Co». Как отмечает Kirk Othmer [4], захват этой технологии составлял цель нескольких научных разведывательных групп стран-победительниц, прибывших в Германию сразу после окончания Второй мировой войны. Отметим, что в составе одной из таких групп был Г.К. Боресков. Впоследствии стадия получения монохлорбензола и стадия его гидролиза были усовершенствованы, и процесс получил название «процесс Рашига»[95].
Гидролиз хлорбензола в отсутствие катализатора протекает при 360—400° С и давлении более 300 ат.
В этих условиях для гидролиза применяются сравнительно концентрированные растворы щелочи (150—200 г/л
NaОН). В присутствии катализатора (медь) эта реакция протекает при 350° С и 200 ат
за несколько минут. Процесс проводят в трубчатых реакторах, причем общую длину труб подбирают с таким расчетом, чтоб при прохождении через них реакционной смеси хлорбензол полностью гидролизовался.
В качестве побочного продукта при гидролизе хлорбензола образуется дифениловый эфир (дифенилоксид):
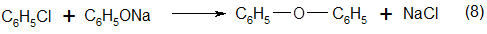
Равновесное количество дифенилового эфира составляет 10% от количества исходного хлорбензола. Если в исходную смесь хлорбензола и водного раствора щелочи специально ввести больше 10% дифенилового эфира, то избыток его гидролизуется в фенол:
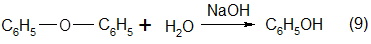
Поэтому дифениловый эфир после отделения от воды добавляют к хлорбензолу, направляемому на гидролиз. Благодаря рециркуляции дифенилового эфира вновь вводимый хлорбензол почти полностью превращается в фенолят натрия и не расходуется на образование эфира.
Промышленный процесс производства фенола заключается в следующем. Насосами высокого давления хлорбензол с растворенным в нем дифениловым эфиром и водный раствор щелочи (мольное соотношение NаОН : С6
Н5
Сl от 2,25 : 1 до 2,5 : 1) подают в трубчатый реактор, где исходная смесь нагревается до 350° С. По выходе из реактора реакционную смесь охлаждают, при помощи дроссельного вентиля снижают давление до атмосферного и отгоняют из смеси дифениловый эфир вместе с парами воды. Фенол выделяют действием двуокиси углерода или соляной кислоты на раствор фенолята и очищают дистилляцией. Расходные коэффициенты на 1 т
фенола: 1,13 т
хлорбензола и 1,24 т
каустической соды. Недостатками метода являются необходимость применения аппаратуры высокого давления, а также относительно высокий расход хлора и каустической соды, вследствие чего в мировой промышленной практике он вытесняется другими способами. Суммарный выход фенола по двум стадиям составляет 70.85%.
Реклама

3.Парофазный каталитический гидролиз хлорбензола (метод Рашига).
Синтез фенола по этому методу обычно проводят в две стадии. В первой стадии получают хлорбензол окислительным хлорированием бензола хлористым водородом в присутствии катализатора:

Хлорирование проводят в паровой фазе над катализатором, состоящим из хлоридов меди и железа на окиси алюминия. Катализатор приготовляют осаждением окиси алюминия из раствора, содержащего хлориды этих металлов. В реакторе поддерживается температура 235 — 245° С. Количественные отношения бензола, хлористого водорода и кислорода составляют примерно 10:2:3. Превращение бензола за один проход 10 — 15%. После удаления бензола, воды и хлористого водорода из хлоридов получают 95 — 98% хлорбензола и 2 — 5% дихлорбензола (количества п-
и о-изомеров относятся как 7:3).

Каталитический гидролиз хлорбензола (вторую стадию процесса Рашига) проводят при атмосферном давлении и 400 — 420° С. В качестве катализаторов используют Са3
(РО4
)2
или смесь фосфатов кальция и меди.
Выделяющийся хлористый водород вновь может быть использован для окислительного хлорирования. Теоретически в этом процессе должны расходоваться только бензол и кислород воздуха; на практике приходится компенсировать потери хлористого водорода.
На 1 т
фенола, получаемого по способу Рашига, расходуется примерно 0,998 т
бензола и 0,172 т
хлористого водорода. Недостатками метода Рашига являются высокие температуры, а также необходимость применения специального коррозионностойкого оборудования [3].
4. Циклогексановый процесс
, разработанный фирмой «Scientific Design Co.», основан на окислении циклогексана в смесь циклогексанона и циклогексанола, которая далее дегидрируется с образованием фенола:

В 60-е годы фирма «Monsanto» в течение нескольких лет использовала этот метод на одном из своих заводов в Австралии, однако в дальнейшем перевела его на кумольный способ получения фенола.
5. В 1961 г. фирмой «DowChemicalofCanada» был реализован процесс через разложение бензойной кислоты
[4] - это единственный способ синтеза фенола, основанный на использовании небензольного сырья:
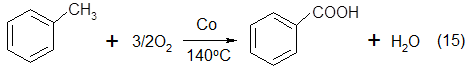
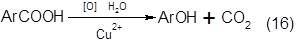
Обе реакции протекают в жидкой фазе. Первая реакция - окисление толуола - использовалась в Германии уже в период Второй мировой войны для получения бензойной кислоты. Реакция протекает в довольно мягких условиях с высоким выходом. Вторая стадия является более трудной вследствие дезактивации катализатора и низкой селективности по фенолу. Полагают, что проведение этой стадии в газовой фазе может сделать процесс более эффективным [5]. [95].
Метод привлек внимание прежде всего потому, что этим способом можно получать фенол из недефицитного толуола вместо бензола. Оказалось также, что метод пригоден для приготовления крезолов из толуиловых кислот и нафтолов из нафтойных кислот. Возможность использования в качестве сырья недефицитные гомологи бензола и нафталина, сравнительно малое количество побочных продуктов, высокая чистота производимых фенолов и использование кислорода воздуха в качестве окисляющего агента — все это вызвало значительный интерес к новому способу производства фенола.
Окислительное декарбоксилирование арилкарбоновых кислот проводится при 200—300°С в присутствии солей двухвалентной меди при подаче в реактор воздуха и водяного пара.
Процесс превращения ароматических карбоновых кислот в фенолы может быть представлен рядом следующих последовательных стадий.
Образование медной соли ароматической карбоновой кислоты:
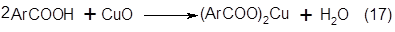
Термическое разложение полученной соли с переходом и с образованием сложного эфира салициловой или смещенной салициловой кислоты:
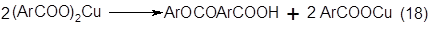
При нагревании солей двухвалентной меди без доступа пара и воздуха исчезает характерное для этих солей синее или зеленое окрашивание, образуются бесцветные соли одновалентной меди. При проведении процесса в более жестких условиях (высокая температуpa, длительное нагревание, недостаток свободной кислоты) образуется элементарная медь.
Это, как и образование одновалентной меди, связано с резким усилением электроноакцепторных свойств меди при повышении температуры.
Регенерация Сu1
и Сu0
.При барботаже воздуха через расплав кислоты, содержащий одновалентную или элементарную медь, эта последняя окисляется до двухвалентного состояния:
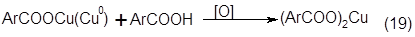
Гидролиз н-декарбоксилирование сложных эфиров. Возможны два направления перехода кислых сложных эфиров в фенолы. В отсутствии водяного пара кислые сложные эфиры декарбоксилируются до эфиров, а последние затем гидролизуются с образованием фенола и исходной кислоты.
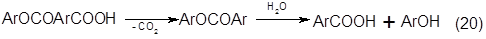
В присутствии водяного пара возможен гидролиз кислых эфиров с образованием исходных арилкарбоновых и оксиарилкарбоновых кислот, а последние декарбоксилируются до фенолов [2].
В настоящее время этот метод используется на практике, хотя его доля в мировом производстве фенола составляет лишь около 5%. Остальные 95% приходятся на кумольный процесс [6].
6. Кумольный процесс
получения фенола включает три стадии: алкилирование бензола в кумол, окисление кумола в гидропероксид кумола и разложение гидропероксида на фенол и ацетон:

Впервые этот способ был разработан в Советском Союзе. Химический маршрут процесса был открыт в 1942 г. группой талантливых химиков, в которую входили П.Г. Сергеев, Р.Ю. Удрис и Б.Д. Кружалов. В это время они были репрессированы и работали в специальной весьма хорошо оборудованной лаборатории, которая одновременно служила и местом заключения.
В 1949 году в г. Дзержинске Горьковской области был введен в действие первый в мире кумольный завод. В 1951 г. в связи с успешным пуском завода большой группе советских ученых и инженеров была присвоена высшая награда страны. Сталинская премия. Драматическая история создания кумольного процесса в СССР описана в ряде статей, опубликованных в 80-е годы [10].
В 1947 г. Сергееву, Удрису и Кружалову были выданы авторские свидетельства СССР [8]. К сожалению, их открытие не получило мировой известности. На Западе кумольный метод был разработан в конце 40-х годов и отчасти известен как процесс Хока [9] - по имени немецкого ученого, позднее независимо открывшего кумольный путь синтеза фенола. В промышленном масштабе этот метод стал впервые использоваться в США в начале 50-х годов [4]. С этого времени на многие десятилетия кумольный процесс становится образцом химических технологий во всем мире.
В настоящее время производство фенола достигло около 6 млн. тонн в год и продолжает быстро расти. Как таковой, фенол практически не используется. Но благодаря тому, что его молекула включает два умеренно реакционноспособных фрагмента (ароматическое кольцо и ОН-группа), каждый из которых может быть вовлечен в дальнейшие превращения [10], фенол является основой для синтеза многих важных химических продуктов. [95].
При окислении углеводородов гидропероксиды образуются по радикально-цепному механизму. Ингибиторы (фенол, олефины, сернистые соединения) сильно тормозят процесс, приводя к появлению индукционного периода, поэтому исходные углеводороды должны быть тщательно очищены от нежелательных примесей. Так, изопропилбензол, полученный алкилированием в присутствии твердого фосфорнокислого катализатора, не пригоден для окисления. Снятию индукционного периода и ускорению реакции на ее начальных стадиях способствует добавление в исходное сырье гидропероксида. Соли металлов переменной валентности, являющиеся обычными катализаторами гомогенного окисления, разлагают гидропероксиды и поэтому не применяются, хотя в отдельных случаях их небольшие добавки ускоряют реакцию. Такой же эффект оказывает металлическая медь, если ее использовать в виде стружек. Иногда медь эффективна, даже если она присутствует в материале аппаратуры. В некоторых исследованиях сообщается о катализе реакции солями металлов постоянной валентности (натрий, калий, магний).
При получении гидропероксидов всегда образуются побочные продукты, главным образом спирты и в меньшем количестве — кетоны. Так, при окислении кумола получаются диметилфенилкарбинол и ацетофенон, причем характер кинетических кривых показывает, что спирт является последовательным продуктом превращения гидропероксида, а кетон образуется параллельно с ним из пероксидного радикала:
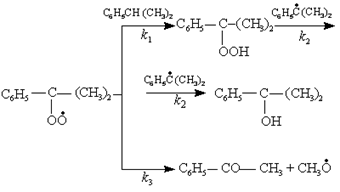 (24) (24)
Кроме свободно-радикального пути расщепления, алкилароматические гидропероксиды способны к распаду под влиянием кислотных и щелочных катализаторов. В присутствии уже небольшого количества сильной кислоты (например, 0,1 % H2
SO4
) гидропероксиды распадаются с образованием фенолов и карбонильных соединений. Реакция протекает по сложному механизму ионного типа с промежуточным возникновением положительных ионов:
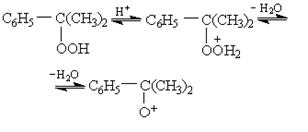 (25) (25)
Образовавшийся ион перегруппировывается с миграцией фенильной группы от углерода к кислородному атому, дальнейшие превращения приводят к образованию фенола и ацетона:
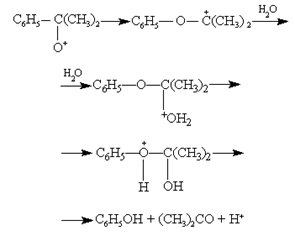 (26) (26)
При другом строении втор
-алкильной группы образуются гомологи ацетона (метилэтилкетон и др.), а из гидропероксидов н
-алкилбензолов — ацетальдегид и его гомологи.
Побочные продукты окисления, содержащиеся как примеси к гидропероксиду (особенно алкилфенилкарбинолы), тоже чувствительны к кислотному катализу. Например, при расщеплении гидропероксида кумола диметилфенилкарбинол отщепляет воду, образуя α-метилстирол, и алкилирует фенол с образованием кумилфенола. Кроме того, α-метилстирол частично димеризуется:
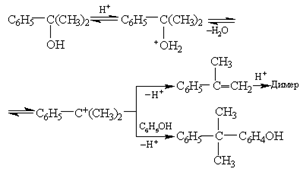 (27) (27)
Получается также небольшое количество смол сложного строения. При повышении концентрации кислоты и температуры становятся возможными кислотно-каталитические превращения ацетофенона и ацетона, например, по типу реакций альдольной конденсации с последующим отщеплением воды.
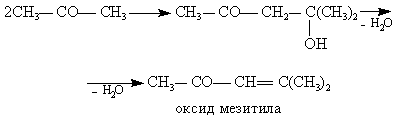 (28) (28)
В кинетическом отношении кислотное разложение гидропероксидов характеризуется очень высокой скоростью, причем практически полное превращение в присутствии 0,05–1 масс. % H2
SO4
(в расчете на гидропероксид) при 50–60 °С достигается за 2–3 мин. Реакция тормозится водой и ускоряется образующимся фенолом, имея первые порядки по кислотному катализатору и гидропероксиду. Вместо серной кислоты в качестве катализаторов можно использовать катионообменные смолы.
Ввиду высокой скорости процесса при его промышленной реализации очень важен эффективный отвод большого количества выделяющегося тепла: 2080 кДж на 1 кг гидропероксида. Для этой цели применяют, прежде всего, разбавители, которыми являются продукты реакции или ацетон.
Один из методов проведения реакции состоит в применении проточно-циркуляционной установки, когда выделяющееся тепло снимают в трубчатом реакторе за счет охлаждения его водой. Реакционную смесь по выходе из реактора частично отводят на дальнейшую переработку, но основное количество направляют на рециркуляцию: добавляют кислоту-катализатор и в насосе смешивают с исходным гидропероксидом. При такой системе время контакта лимитируется теплоотводом и является завышенным. Кроме того, рециркуляция смеси ведет к повышенному выходу побочных веществ. Так, на 1 т фенола получается 100–150 кг отходов, в том числе 15–20 кг α-метилстирола, 40–50 кг димера и смол, 5–10 кг ацетофенона, 30 кг кумилфенола и т. д. Хотя оксида мезитила образуется немного, он существенно затрудняет очистку фенола.
Другой способ кислотного разложения гидропероксидов состоит в проведении реакции в растворе ацетона и отводе тепла за счет его испарения. Ацетон конденсируют в обратном холодильнике и возвращают в реактор, который можно секционировать поперечными перегородками. Это (наряду с уменьшением концентрации фенола в растворе и времени контакта) снижает выход побочных веществ.
Современный процесс получения фенола и ацетона включает 10 основных стадий: получение изопропилбензола (ИПБ); окисление его до гидропероксида кумола (ГПК); концентрирование продуктов окисления, содержащих 20–40 % гидропероксида, путем отгонки под вакуумом (1–3 стадии) непрореагировавшего кумола; разложение ГПК в среде фенола, ацетона и кумола в присутствии серной кислоты до фенола, ацетона, α-метилстирола и побочных продуктов (так называемой «фенольной смолы»); нейтрализация серной кислоты и выведение из системы Na2
SO4
и NaHSO4
; разделение полученных продуктов до индивидуальных веществ; гидрирование α-метилстирола до кумола; очистка фенола-сырца от микропримесей с использованием кислотных катионитов; очистка ацетона от микропримесей с использованием щелочей; частичный термический крекинг побочных продуктов производства (рис. 2). Окисление проводится в тарельчатой реакционной колонне, снабженной холодильниками; при их помощи поддерживают температуру жидкости от 120 °С на верхней тарелке до 105 °С в кубе. Воздух, предварительно очищенный от загрязнений и механических примесей и подогретый, подают в нижнюю часть колонны под давлением 0,4 МПа. Свежий и оборотный изопропилбензол, к которому добавлен ГПК, инициирующий начальную стадию окисления, подают в подогреватель, а оттуда на верхнюю тарелку реактора.
Воздух движется противотоком к жидкости, барботируя через нее на тарелках колонны. При этом он увлекает с собой пары изопропилбензола и летучих побочных продуктов (муравьиная кислота, формальдегид), которые конденсируются в холодильнике. Оставшийся воздух выводят в атмосферу, а конденсат отмывают от муравьиной кислоты водной щелочью в промывателе-сепараторе. Углеводородный слой сливают в сборник, а водный слой рециркулируют на промывку, сбрасывая в конечном счете в канализацию.
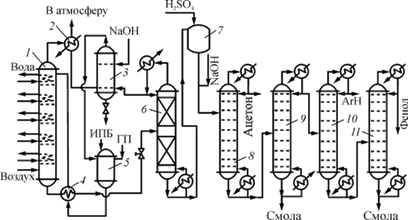
Рис. 2.
Технологическая схема кумольного метода получения фенола и ацетона: 1
— реакционная колонна; 2
— холодильник; 3
— промыватель-сепаратор; 4
— теплообменник; 5
— сборник; 6
, 8–11
— ректификационные колонны; 7
— узел кислотного разложения ГПК
Оксидат из нижней части колонны отдает свое тепло изопропилбензолу в теплообменнике, дросселируется до остаточного давления 4 кПа и поступает на вакуум-ректификацию для концентрирования гидропероксида. Отгонку изопропилбензола ведут в насадочной ректификационной колонне непрерывного действия, снабженной конденсатором-дефлегматором. Применение вакуума обусловлено термической нестабильностью гидропероксида. Часть конденсированного изопропилбензола возвращают из конденсатора-дефлегматора на орошение колонны, а остальное количество выводят в сепаратор, промывают щелочью и снова направляют на окисление.
Кубовая жидкость из колонны содержит 70–75 % гидропероксида (ГП), а также побочные продукты окисления и остатки изопропилбензола. Путем дополнительной вакуум-ректификации при остаточном давлении 665 Па повышают концентрацию гидропероксида до 88–92 %. Следующую стадию (кислотное разложение гидропероксида) осуществляют одним из двух описанных выше методов [3].
Недостатки кумольного метода.
Несмотря на прекрасно отлаженную технологию и длительный опыт эксплуатации, кумольный метод имеет ряд недостатков. Прежде всего это наличие взрывоопасного промежуточного соединения (гидропероксид кумола), а также многостадийность метода, что требует повышенных капитальных затрат и делает труднодостижимым высокий выход фенола в расчете на исходный бензол. Так, при выходе полезного продукта 95% на каждой из трех стадий итоговый выход составит лишь 86%. Приблизительно такой выход фенола и дает кумольный метод в настоящее время.
Но самый важный и принципиально неустранимый недостаток кумольного метода связан с тем, что в качестве побочного продукта образуется ацетон. Это обстоятельство, которое первоначально рассматривалось как сильная сторона метода, становится все более серьезной проблемой, поскольку ацетон не находит эквивалентного рынка сбыта. В 90-х годах эта проблема стала особенно ощутимой после создания новых способов синтеза метилметакрилата путем окисления углеводородов С4, что резко сократило потребность в ацетоне. Об остроте ситуации говорит тот факт, что в Японии разработана технология, предусматривающая рецикл ацетона. С этой целью к традиционной кумольной схеме добавляются еще две стадии - гидрирование ацетона в изопропиловый спирт и дегидратация последнего в пропилен [11]:
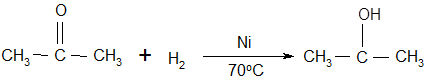 (29) (29)
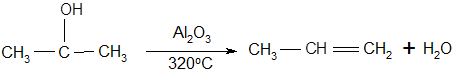 (30) (30)
Образующийся пропилен снова возвращают на стадию алкилирования бензола. В 1992 году фирма «Mitsui» пустила крупное производство фенола (200 тыс. т/год), основанное на этой пятистадийной кумольной технологии.
Предлагаются также другие сходные модификации кумольного метода, которые позволили бы смягчить проблему ацетона. Однако все они приводят к значительному усложнению технологии и не могут рассматриваться как перспективное решение проблемы. Поэтому исследования, ориентированные на поиск новых путей синтеза фенола, которые основывались бы на прямом окислении бензола, в последнее десятилетие приобрели особенно интенсивный характер. Работы ведутся главным образом в следующих направлениях: окисление молекулярным кислородом, окисление моноатомными донорами кислорода и сопряженное окисление.
Отметим, что аналогичные подходы применяются и для поиска эффективных способов селективного окисления низших парафинов, включая метан.
2. Поиск новых путей синтеза фенола
Окисление молекулярным кислородом.
Прямое окисление бензола молекулярным кислородом представляется наиболее привлекательным методом получения фенола:
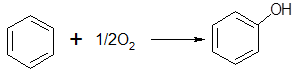 (31) (31)
Однако это на первый взгляд самое простое и очевидное решение проблемы оказалось чрезвычайно трудной задачей. Работы по окислению бензола с помощью О2
начались еще до того, как в 1865 г. Кекуле предложил структурную формулу бензольного кольца [12]. С тех пор многочисленные попытки найти эффективный путь для проведения этой реакции не прекращаются. Окисление бензола ведут как в жидкой, так и в газовой фазах, при низком и высоком давлениях, в отсутствие и в присутствии разнообразных катализаторов [13]. Несмотря на отдельные успехи, результаты этих работ пока далеки от практического применения.
Начиная с 80-х годов, значительные усилия предпринимаются для проведения этой реакции в жидкой фазе с использованием в качестве катализаторов различных комплексов переходных металлов, среди которых наибольшую активность проявляют соединения Pd и Cu [14,15]. Однако после нескольких оборотов реакция, как правило, прекращается вследствие деградации катализатора.
Моноатомные доноры кислорода.
Более успешные результаты дает применение в качестве окислителей так называемых моноатомных доноров кислорода в виде различных кислородсодержащих молекул. Среди таких молекул наибольшее внимание привлекает пероксид водорода:
 (32) (32)
Окисление с помощью Н2
О2
проводят в присутствии солей и комплексов переходных металлов, в том числе инкапсулированных в матрице цеолита [16,17]. Показана возможность проведения этой реакции в условиях межфазного катализа [18].
Исследования в данной области приобрели особенно интенсивный характер после открытия цеолитов состава Ti-Si (TS-1) и их уникальных свойств в реакциях жидкофазного окисления с помощью пероксида водорода [19]. На этой основе фирмой «Enichem» разработан промышленный процесс получения гидрохинона и пирокатехина путем гидроксилирования фенола. Вслед за цеолитами TS-1 были опробованы разнообразные цеолитные системы, содержащие как титан, так и другие переходные металлы [20-22]. Однако, в отличие от окисления фенола, ароматическое кольцо которого активировано присутствием ОН-группы, окисление бензола протекает менее активно и со значительно меньшей селективностью по пероксиду водорода вследствие его побочного разложения на кислород и воду.
Следует отметить, что в любом случае реакция (16) едва ли перспективна для практического использования из-за высокой стоимости Н2
О2
(по сравнению со стоимостью фенола).
Помимо пероксида водорода, в исследовательских целях применяется ряд других, более сложных и дорогостоящих монокислорододонорных окислителей, таких как перкислоты, иодозобензол и т.д. Однако для окисления бензола они практически не используются.
Более вероятным окислителем для бензола с практической точки зрения представляется азотная кислота, которая впервые была использована для этой цели в 1925 г. В более поздних работах было показано, что эффективными катализаторами этой реакции являются оксидные системы на основе V2
O5
и MoO3
[23]:
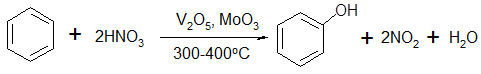 (33) (33)
Окисление протекает в газовой фазе в присутствии паров воды при температуре 300-400 0
С. Конверсия бензола достигает 52% при селективности по фенолу более 90%. Образующийся диоксид азота может быть окислен до HNO3
и вновь использован для окисления бензола.
Заслуживает упоминания способ окисления бензола, основанный на реакции с сульфатом двухвалентной меди в качестве окислителя:
 (34) (34)
Реакция протекает за счет восстановления Cu2+
в Cu+
или даже в Cu0
. Первые патенты в этой области появились в 60-е годы [24]. В последнее время интерес к этой реакции возобновился [25].
Есть сообщение [26] об использовании воды в качестве окислителя бензола:
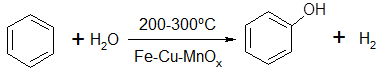 (35) (35)
По данным этой работы, при нормальном давлении и температуре 200-300 0
С в присутствии смешанных оксидов Fe, Cu и Mn достигается конверсия бензола 15% с селективностью по фенолу 97%. Однако этот результат требует серьезной проверки. Вероятнее всего, образование фенола здесь связано не с реакцией (19), протекание которой с таким выходом фенола невозможно в силу термодинамических ограничений, а обусловлено окислением бензола за счет кислорода катализатора.
К рассматриваемому классу реакций относится и реакция окисления бензола в фенол с помощью закиси азота, на основе которой создан новый одностадийный способ получения фенола (см. ниже).
Сопряженное окисление.
Пероксид водорода может непосредственно образовываться в реакционной системе in situ
и тут же расходоваться на окисление субстрата. Этот подход рассматривается как одно из наиболее перспективных направлений не только для окисления ароматических соединений, но и многих других углеводородов, включая парафины [27,28]. Использование пероксида по мере его образования из Н2
и О2
позволяет значительно увеличить селективность полезного использования H2
O2
. Это типичный пример сопряженного процесса, протекающего с участием вещества-«жертвы» [29]:
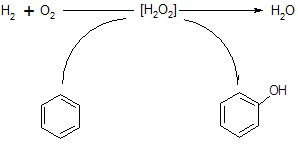 (36) (36)
Напомним, что механизм сопряжения заключается в том, что промежуточное вещество, образующееся при протекании первичной реакции, частично используется для осуществления вторичной реакции, которая сама по себе в этих условиях не происходит. В данном случае первичной реакцией является окисление водорода в H2
O через промежуточное образование H2
O2
, а вторичной реакцией - окисление бензола в фенол.
Работы в этом направлении интенсивно ведутся во многих лабораториях мира с использованием разнообразных катализаторов и приемов [27,30]. Наиболее эффективные катализаторы включают платину или другой благородный металл, который вместе с оксидом ванадия наносится на силикагель. На таком катализаторе в растворе уксусной кислоты бензол почти со 100%-ной селективностью окисляется в фенол. Однако селективность реакции по водороду все еще остается невысокой и составляет 10-15% (это значит, что на образование 1 моль фенола расходуется 7-8 моль водорода).
Сопряженное окисление водорода и бензола может протекать также в газовой фазе при повышенной температуре [31].
Помимо окисления водорода, в качестве первичной реакции может выступать процесс окисления других молекул, например монооксида углерода [32]:
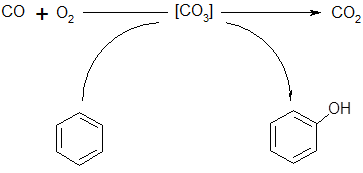 (37) (37)
Сопряжение может быть организовано также в сочетании с окислением восстановленной формы металла в растворах:
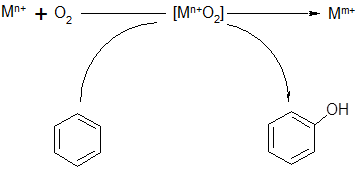 (38) (38)
Окисление бензола здесь стимулируется окислительными превращениями металлов: Fe2+
® Fe3+
, Sn2+
® Sn4+
[33], Cu+
® Cu2+
[34], Zn0
® Zn2+
[35]. Иногда, как в случае использования цинковой пыли, для протекания реакции требуется введение в систему дополнительного катализатора, в качестве которого могут выступать соли переходных металлов.
Недостаток сопряженных реакций состоит в трудности подбора таких условий, которые были бы оптимальными как для образования сопрягающего промежуточного соединения в первичной реакции, так и для его эффективного использования во вторичной реакции.
В последние годы широкое развитие получают работы по исследованию реакций сопряженного окисления углеводородов в электрохимических ячейках, представляющих собой реакторы типа топливных элементов [36-38]. Проведение реакций в таких устройствах открывает дополнительную возможность для управления реакцией посредством регулирования электрического тока, пропускаемого через ячейку.
В заключение данного раздела отметим, что реакция окисления бензола в фенол, вероятно, является рекордсменом по разнообразию подходов, количеству испытанных каталитических систем, а также по объему усилий, предпринимаемых исследователями для осуществления этой реакции. Сравниться может только реакция окисления метана в метанол, которая является столь же простой на первый взгляд и столь же трудной для реального воплощения, как и окисление бензола. Осуществление этих реакций, несомненно, останется заветной мечтой и для химиков XXI века.
3. Открытие цеолитных катализаторов для окисления бензола закисью азота
Термодинамически N2
O - неустойчивая эндотермическая молекула, свободная энергия образования которой равна 103,6 кДж/моль. Однако кинетически закись азота весьма стабильна, ее некаталитическое разложение становится заметным лишь при температуре выше 600 0
С.
В 1983 году Ивамото с сотруд. впервые использовал закись азота для окисления бензола в фенол [39]:
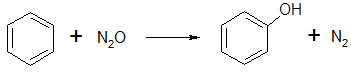 (39) (39)
Эта реакция позволила выйти на принципиально новый уровень селективности по фенолу. При температуре 550 0
С и добавлении в реакционную смесь 30% H2
O селективность по фенолу на нанесенных ванадиевых катализаторах достигала 71% при конверсии бензола 10%. Этот результат вызвал со стороны ученых оживленные отклики, в которых особо подчеркивалась экологическая безопасность нового процесса и давались весьма оптимистические оценки относительно его практической перспективы [40,41]. Однако пилотные испытания, проведенные в реакторе с кипящим слоем катализатора, не подтвердили эти ожидания. Селективность ванадиевых катализаторов оказалась недостаточно высокой для практического применения процесса.
Тем не менее, работа Ивамото и сотруд. [39] стимулировала поиск новых более эффективных катализаторов. Испытания огромного числа каталитических систем выявили чрезвычайно высокую требовательность реакции окисления бензола в фенол закисью азота. Катализаторы оказывались либо неактивными, либо неселективными [42].
В 1988 г. три группы исследователей - Сузуки с соавт. [43] (Токийский технологический институт), Губельманн и Тирель [44] (фирма «Rhone Poulenc») и Харитонов с соавт. (Институт катализа СО РАН) независимо показали, что лучшими катализаторами этой реакции являются цеолиты ZSM-5. На цеолитах реакция протекает при значительно более низкой температуре и, что особенно важно, с селективностью по фенолу, близкой к 100%. Несмотря на сравнительно небольшой выход фенола, полученный в этих работах, было ясно, что найдена перспективная основа для создания новых катализаторов. Дальнейшие исследования [45-55] позволили значительно улучшить каталитические свойства цеолитов и расширить круг катализируемых ими реакций, включая окисление закисью азота различных производных бензола. В настоящее время на этой основе фирмой «Solutia» (бывшая фирма «Monsanto», США) и Институтом катализа СО РАН ведется создание нового фенольного процесса, который успешно прошел пилотные испытания и принят фирмой к промышленному освоению [56].
4. Природа каталитической активности цеолитов
Открытие цеолитных катализаторов, представляющих собой весьма нетипичные системы для окислительного катализа, поставило ряд вопросов фундаментального характера, среди которых вопрос о природе каталитической активности цеолитов вызывает наиболее оживленные дискуссии.
Для объяснения каталитической активности цеолитов были выдвинуты две гипотезы. Одна из них основана на традиционных представлениях о цеолитах как кислотных катализаторах, обладающих сильными бренстедовскими и льюисовскими кислотными центрами. Объяснение, исходящее из представлений о бренстедовских центрах, предполагает их способность протонировать молекулы закиси азота, что приводит к образованию гидроксил-катионов, которые далее атакуют молекулы бензола:
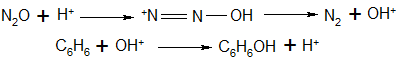 (40) (40)
Эта гипотеза впервые была высказана в работе [42] и затем, с теми или иными оговорками, принята рядом других авторов [45,47]. Однако специальное исследование, выполненное позднее с привлечением инфракрасной спектроскопии, не подтвердило эту гипотезу [57].
Ряд исследователей связывают активность цеолитов с льюисовскими кислотными центрами, которые образуют координационно ненасыщенные атомы алюминия или галлия, вышедшие из кристаллической решетки [46,48,49,52]. Это объяснение основывается на том, что с увеличением жесткости условий высокотемпературной обработки цеолита, проводимой на воздухе [46,50], в вакууме [57] или в присутствии паров воды [52,58], возрастает как концентрация льюисовских кислотных центров, так и каталитическая активность цеолита в реакции окисления бензола в фенол. Однако авторы этих работ не приводят каких-либо количественных корреляций между кислотностью и каталитической активностью.
В работах Института катализа СО РАН развивается другой подход, основанный на представлениях окислительного катализа. Исходная позиция этого подхода заключается в том, что сама по себе алюмосиликатная матрица цеолита, независимо от её кислотных свойств, неспособна катализировать такую тонкую и трудную реакцию как окисление бензола в фенол. Причина каталитической активности, вероятнее всего, связана с примесью какого-либо переходного металла. Действительно, как выяснилось в дальнейшем, таким металлом оказалось железо - наиболее распространенная примесь, вносимая в основном с реагентами на стадии синтеза цеолита [41].
Роль железа была детально исследована на примере двух специально синтезированных Fe-содержащих цеолитных систем со структурой ZSM-5. Железо вводилось на стадии синтеза цеолитов и в одном случае его включали в Al-Si матрицу [59], а в другом - в чисто силикатную матрицу, синтезированную в отсутствие алюминия [60]. В обоих случаях самые чистые образцы, которые авторам удалось приготовить, принимая максимальные предосторожности против загрязнения железом (0,002-0,003 % масс. Fe), оказались неактивными. Активность появлялась только с введением железа, увеличиваясь по мере роста его концентрации. Это связано с тем, что железо в цеолите образует особые активные центры, названные a-центрами (их природа рассмотрена ниже), которые и обеспечивают протекание каталитической реакции. На рис. 3 показана зависимость концентрации каталитически активных центров от содержания Fe для обеих цеолитных систем. Несмотря на сходный характер, кривые существенно сдвинуты относительно друг друга по оси абсцисс. Так, введение в алюмосиликатную матрицу железа уже на уровне сотых долей процента приводит к образованию значительного количества a-центров, тогда как в случае силикатной матрицы для этого требуется ввести в 10-30 раз большее количество железа. Это говорит о том, что присутствие алюминия способствует образованию a-центров, хотя причина этого пока остается неясной. Одно из объяснений может заключаться в том, что активные комплексы Fe занимают катионнообменные позиции в цеолите и стабилизируются на атомах алюминия [49].
lgC
a [центр/г]
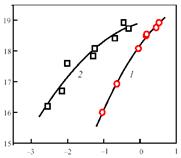
lgC
Fe [% масс.]
Рис. 3. Зависимость концентрации a-центров от концентрации железа в цеолитах ZSM-5 состава Fe.Si (1
) и Fe.Al.Si (2
)
Интересно, что независимо от состава матрицы и количества введенного железа a-центры проявляют удивительную равноценность в своих каталитических свойствах. Это относится как к реакции разложения N2
O [61], так и к реакции окисления бензола в фенол [41]. По отношению к последней реакции такой эффект хорошо виден из рис. 4, на котором приведена зависимость конверсии бензола от концентрации a-центров. Активность всех исследованных образцов описывается единой зависимостью и определяется только концентрацией a-центров. Согласно квантовохимическим расчетам [62], a-центры представляют собой биядерные комплексы железа, которые формируются на стадии выхода Fe из кристаллической решетки цеолита при его термических обработках. Аналогично внерешеточному алюминию, эти комплексы являются координационно ненасыщенными частицами и обладают льюисовской кислотностью. Учитывая сходный механизм образования внерешеточных частиц Fe и Al, естественно ожидать некоторую корреляцию между каталитической активностью и льюисовской кислотностью, что и наблюдали при различных высокотемпературных обработках одного и того же образца цеолита. Однако в случае образцов с различным химическим составом такая корреляция не должна существовать, что действительно и подтверждается [60]. Это объясняется тем, что в разных образцах неодинакова концентрация вне решеточных атомов Al, которые дают основной вклад в льюисовскую кислотность, но не дают вклада в каталитическую активность. Квантовохимические расчеты также не подтверждают каталитическую роль льюисовских центров [63].
Конверсия бензола,%
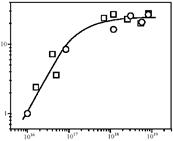
C
a, a-центр/г
Рис. 4. Зависимость конверсии бензола от концентрации a-центров на цеолитах ZSM-5 состава Fe.Si  и Fe.Al.Si и Fe.Al.Si 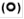 при 350 °С при 350 °С
Каталитическая активность железа в реакциях окисления закисью азота подтверждена в ряде работ других авторов [50, 51, 64].
Интересно, что образование a-центров приводит к качественному изменению в состоянии железа по сравнению с атомами Fe на поверхности оксида Fe2
O3
. В составе a-центров атомы Fe теряют способность активировать молекулярный кислород, но приобретают повышенную способность к активации молекул N2
O [61]. Это ярко проявляется в реакции окисления бензола с помощью этих молекул (табл. 1). Так, в присутствии закиси азота конверсия бензола составляет 27% при 350 0
С, тогда как в присутствии О2
. только 0,3% при 500 0
С. Более того, при этом изменяется направление реакции: если при действии N2
O с высокой селективностью образуется фенол, то в случае О2
регистрируются лишь продукты полного окисления. На оксиде железа ни один из окислителей не приводит к образованию фенола.
5. Специфика действия N2
O как окислителя
Специфика закиси азота как окислителя - это один из наиболее важных вопросов, возникающих при исследовании реакции окисления бензола в фенол. Преимущества закиси азота по сравнению с O2
наглядно демонстрируют результаты, представленные в табл. 2. Естественно предположить, что столь сильное влияние природы окислителя связано с существенным изменением в состоянии поверхностного кислорода. Это предположение стимулировало постановку ряда работ по исследованию механизма реакции разложения закиси азота на цеолитах FeZSM-5, поскольку именно за счет этой реакции осуществляется поставка кислорода на поверхность катализатора.
Таблица 1
Исследования позволили установить, что разложение N2
O протекает на a-центрах цеолита с образованием новой формы поверхностного кислорода, условно названной a-кислородом [65,66]. В низкотемпературной области реакция образования a-кислорода носит стехиометрический характер:
N2
O+ ( )a
= (O)a
+ N2
(41)
На рис. 5 приведена кинетическая кривая низкотемпературного разложения N2
O в статической установке. Видно, что протекание реакции сопровождается выделением в газовую фазу только N2
, тогда как образующийся кислород заполняет a-центры и остается полностью связанным на поверхности. По мере заполнения a-центров реакция прекращается.
Давление, мм.рт.ст.
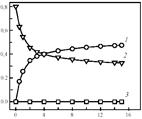
Время, мин
Рис. 5. Изменение парциального давления N2
(1
), N2
O (2
) и O2
(3
) при разложении закиси азота на цеолите FeZSM-5 при 250 °С
До температуры 3000
С a-кислород термически устойчив. Выше этой температуры он необратимо десорбируется в газовую фазу и реакция разложения N2
O приобретает характер обычного каталитического процесса.
Для проведения дальнейших количественных исследований важно знать концентрацию a-кислорода. Существует несколько методов, позволяющих надежно определить данную величину. Это может быть выполнено как путем непосредственного измерения количества выделившегося N2
(или израсходованного N2
O) в ходе реакции (41) или путем десорбции Оa
в газовую фазу при нагревании образца выше 300 0
С. Кроме того, удобно уже использовать реакцию изотопного обмена, в которую a-кислород легко вступает при комнатной температуре:
(16
O)a
+ 18
O2
= (18
O)a
+ 16
O18
O(42)
В состоянии равновесия доли изотопа 18
О в поверхностном a-кислороде и в кислороде газовой фазы равны, что позволяет рассчитать количество Оa, вступившего в обмен. Собственный кислород цеолита инертен в отношении изотопного обмена и не мешает измерению концентрации a-кислорода.
Исходя из максимального количества a-кислорода, которое может быть «посажено» на данном образце цеолита (уравнение 41), рассчитывается концентрация a-центров С
a в предположении, что один атом кислорода занимает один центр. Значения С
a, определенные разными методами, хорошо согласуются между собой [66]. Для наиболее активных образцов с достаточно высоким содержанием железа величина С
a может достигать 100 мкмоль/г.
Изучению свойств a-кислорода посвящено значительное число работ, результаты которых рассмотрены в обзоре [67]. Особенно следует отметить низкую энергию связи Оa с поверхностью и очень высокую реакционную способность, благодаря которой он способен уже при комнатной температуре окислять различные углеводороды.
6. Стехиометрическая реакция бензола с
a-кислородом
Установление специфической способности закиси азота генерировать a-форму поверхностного кислорода подводит нас к главному вопросу относительно механизма реакции, а именно, к вопросу об участии a-кислорода в образовании фенола.
Согласно общепринятой точке зрения, которая особенно ясно сформулирована в работах школы Борескова применительно к классическим оксидным катализаторам [69], поверхностный кислород, участвующий в парциальном окислении, не должен обладать низкой энергией связи с поверхностью и не должен иметь высокую реакционную способность, что находится в явном противоречии со свойствами a-кислорода. Поэтому идея соотнести образование фенола с генерацией a-кислорода требует серьезного экспериментального подтверждения.
Следует отметить, что идентификация поверхностных форм кислорода, принимающих участие в реакциях окисления, представляет собой очень трудную задачу. В условиях катализа при повышенных температурах происходят быстрые взаимные превращения различных форм кислорода [70,71], что делает результаты малоинформативными. При пониженной температуре, когда таких превращений нет, идентификацию поверхностных форм кислорода обычно также не удается провести либо из-за их низкой активности, либо из-за малой концентрации. В случае же a-кислорода ситуация кажется уникальной - высокая активность и большая концентрация этой формы кислорода, которую можно регулировать (путем введения Fe) в пределах нескольких порядков. Это обстоятельство позволило нам простым и надежным способом ответить на вопрос об участии a-кислорода в реакции окисления бензола [66].
Сущность экспериментов заключалась в титровании a-кислорода бензолом, которое проводилось по схеме, включающей «посадку» кислорода на a-центр, его взаимодействие с бензолом при комнатной температуре и экстракцию продукта метанолом:
N2
O + ( )a
 (O)a
+ N2 (O)a
+ N2
C6
H6
+ (O)a
 (C6
H5
OH)a
(43) (C6
H5
OH)a
(43)
(C6
H5
OH)a
C6
H5
OH+ ( )a
Как показали исследования, в пределах точности эксперимента выход фенола близок к теоретическому. Других продуктов реакции не обнаружено. Полученные результаты, несомненно, говорят об участии a-кислорода в образовании фенола. Позднее этот вывод был подтвержден дополнительными экспериментами с использованием a-кислорода, обогащенного изотопом 18
О.
7. Биомиметические свойства
a-кислорода
Факт образования фенола при комнатной температуре в рассматриваемой реакции наводит на мысль о вероятном сходстве a-кислорода с активным кислородом монооксигеназ, для которых гидроксилирование ароматических соединений является типичной реакцией [72]. Напомним, что монооксигеназами называют ферменты, способные при комнатной температуре селективно вести окислительные реакции, в ходе которых один атом кислорода, переведенный в активное состояние, присоединяется к неактивированной молекуле углеводорода-субстрата, давая ОН-содержащее соединение, а второй - расходуется на образование воды. Активация кислорода является наиболее трудной проблемой биомиметической химии, особенно в случае моделирования так называемых метанмонооксигеназ. Биядерные Fe-содержащие центры метанмонооксигеназ способны генерировать кислородные частицы, которые по активности значительно превосходят кислород других монооксигеназ. Вследствие этого, помимо способности гидроксилировать ароматические и другие соединения, метанмонооксигеназы обладают уникальной возможностью гидроксилировать даже метан - наиболее инертную органическую молекулу.
Изучение реакции a-кислорода с метаном [66,73] с использованием той же трехстадийной схемы титрования, что и в вышеприведенном случае с бензолом («посадка» Oa
, его взаимодействие с метаном при комнатной температуре и экстракция продукта) показало, что реакция протекает очень быстро, с образованием только метанола, количество которого соответствует количеству прореагировавшего метана.
Аналогичные результаты были получены для реакций с этаном, пропаном и рядом других углеводородов: окисление a-кислородом при комнатной температуре приводит к образованию тех же продуктов, что и при окислении с помощью монооксигеназ.
Для установления более глубокой аналогии a-кислородного окисления с биологическим окислением важно сравнить не только продукты, но и механизм, по которому они образуются в обоих случаях. Удобным инструментом для этой цели является измерение кинетического изотопного эффекта (КИЭФ). Известно, что биологическое окисление метана протекает с большим изотопным эффектом (от 5 до 12), тогда как в случае окисления бензола кинетический эффект не наблюдается [74]. Для сопоставления механизмов обсуждаемых процессов в работе [75] было проведено измерение КИЭФ для реакций окисления метана и бензола a-кислородом.
Молекулы метана CH2
D2
, которые использовались в работе [75] могут реагировать либо по C-H, либо по C-D связи, давая две изотопные разновидности метанола:
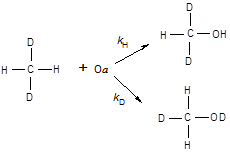 (44) (44)
Экстрагированный с поверхности катализатора метанол был проанализирован методом ЯМР и на основании ЯМР-спектров рассчитаны величины изотопного эффекта. Оказалось, что в зависимости от температуры КИЭФ меняется от 5,5 при - 50 0
С до 1,9 при +100 0
С.
Высокие значения КИЭФ убедительно указывают на тот же механизм реакции, что и при окислении с помощью метанмонооксигеназ. В обоих случаях лимитирующая стадия реакции включает разрыв связи С-Н.
При взаимодействии a-кислорода с бензолом также образуются две изотопные разновидности фенола, содержащие ОН- и OD-группы. Результаты измерения КИЭФ и здесь показали сходство с биологическим процессом: как и в случае действия монооксигеназ, окисление бензола a-кислородом протекает без изотопного эффекта. Это значит, что лимитирующая стадия реакции не включает разрыв C-H связи и, вероятно, протекает через промежуточное образование ареноксида [75]:
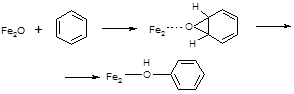 (45) (45)
Таким образом, a-кислород во многих отношениях аналогичен активному кислороду метанмонооксигеназ: он координирован на комплексах Fe, обладает высокой реакционной способностью и направляет окисление по механизму того же типа.
8. Активное состояние железа в цеолитной матрице
При изучении активного состояния железа, входящего в состав a-центров, возникают два вопроса: о локализации a-центров и о структуре a-центров.
В общем случае железо в матрице цеолита может быть локализовано в трех состояниях [76]:
1) как изолированные ионы в тетраэдрических позициях кристаллической решетки (вместо Si);
2) как изолированные ионы или малые кластеры внутри микропористого пространства цеолита;
3) как частицы оксида на внешней поверхности кристаллов цеолита.
Было проведено несколько специальных исследований [77-80], чтобы понять, какому из этих состояний соответствуют a-центры. Результаты этих исследований позволили надежно исключить первое и третье состояния, так что a-центры, включающие Fe, могут находиться только во внутри-кристаллическом пространстве цеолита.
Вопрос о структуре a-центров более сложный. Анализ результатов, проведенный в работе [81], указывает на то, что a-центры, вероятно, представляют собой биядерные комплексы железа. В пользу этого вывода свидетельствуют адсорбционные измерения, результаты ЭПР [61], квантовохимические расчеты [62,68] и данные мессбауэровской спектроскопии [82,83]. Отметим, что последний метод широко применяется для анализа Fe в составе метанмонооксигеназ. Использование мессбауэровской спектроскопии по отношению к цеолиту FeZSM-5 позволило обнаружить удивительное согласие между спектральными параметрами Fe в цеолите и в составе биядерных центров метанмонооксигеназ [81].
Косвенным доводом в пользу биядерного строения a-центров можно считать то обстоятельство, что биядерные оксо- (или гидроксо-) комплексы металлов, по-видимому, широко используются в природе для генерации высокоактивных частиц кислорода. Так, предполагается, что ферменты, осуществляющие окисление воды, а также их искусственные модели содержат биядерные комплексы в качестве функциональных единиц [84].
Тем не менее, имеющихся экспериментальных данных пока недостаточно, чтобы считать биядерную структуру a-центров окончательно доказанной. Полностью нельзя исключить и моноядерный вариант структуры.
Как показали мессбауэровские измерения, «посадка» и снятие Oa сопровождается обратимыми окислительно-восстановительными переходами железа
в составе FeZSM-5.
Состояние Fe в матрице цеолита ZSM-5 является предметом изучения многих исследовательских групп [85-90]. В ряде работ также приводятся аргументы в пользу образования биядерных комплексов железа [85,86,88]. Однако следует отметить, что в этих работах, как правило, имеют дело с более высокими концентрациями Fe.
9. Новый фенольный процесс. Технологические аспекты
Разработка нового фенольного процесса, основанного на прямом окислении бензола закисью азота, ведется в рамках сотрудничества между Институтом катализа СО РАН и американской фирмой «Solutia», которая является новой химической компанией, отделившейся от фирмы «Monsanto» около двух лет назад. «Solutia» принадлежит к числу крупнейших в мире производителей адипиновой кислоты (полупродукта для синтеза найлона 6,6), что в значительной мере и определило её первоначальный интерес к каталитической химии закиси азота.
Технологическая схема получения адипиновой кислоты (рис. 4) включает следующие стадии: гидрирование бензола в циклогексан, окисление циклогексана кислородом воздуха в смесь циклогексанола и циклогексанона и, наконец, дальнейшее окисление этой смеси в адипиновую кислоту с помощью азотной кислоты. Последняя стадия дает большое количество отходов закиси азота, которая образуется приблизительно в мольном соотношении 1:1 с адипиновой кислотой. В настоящее время закись азота уже не рассматривается как безобидный «веселящий газ». Она обладает сильным парниковым эффектом [92], который (в расчете на моль) в 160 раз превосходит эффект СО2
, главного «парникового газа». Кроме того, имея большое время жизни, N2
O достигает верхних слоев атмосферы, где способствует разрушению озонового слоя Земли. Если учесть, что человек ежегодно выбрасывает в атмосферу более 10 млн. тонн N2
O (из них около 10% связано с производством адипиновой кислоты), то можно понять, почему закись азота вызывает растущее беспокойство экологов и почему в ряде стран принимаются законы, ограничивающие выбросы N2
O.
В этой ситуации фирма «Solutia» предложила идею использовать отходы закиси азота для окисления бензола в фенол и включить эту реакцию как ключевую стадию в модифицированную схему адипиновой кислоты (см. рис. 6). По этой схеме бензол сначала окисляется в фенол с помощью N2O и лишь затем идет на стадию гидрирования. Закись азота, образующаяся на последней стадии, возвращается в начало процесса, замыкая, таким образом, свой технологический цикл.
Новый фенольный процесс основан на использовании Fe-содержащих цеолитов ZSM-5, и по имени a-кислорода назван AlphOx-процессом. Окисление бензола осуществляется в простом адиабатическом реакторе, что имеет немаловажное значение с точки зрения экономичности процесса.
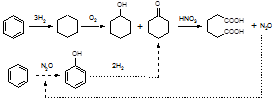
Рис.6. Существующая (сплошные стрелки) и модифицированная (пунктирные стрелки) схемы получения адипиновой кислоты
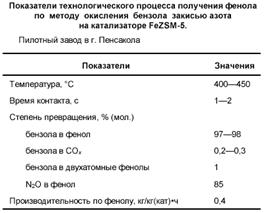
В табл. 2 приведены типичные технологические показатели процесса на пилотном заводе, построенном фирмой в г. Пенсакола (США) [93].
Производство функционирует в режиме непрерывной рециркуляции газовых и жидких потоков с выделением из продуктов чистого фенола. Процесс обладает высокой селективностью в расчете как на бензол, так и на закись азота. Его производительность (400 г фенола с 1 кг катализатора в час) является высоким показателем для реакций селективного окисления. Кроме того, AlphOx-процесс обладает рядом других преимуществ по сравнению с кумольным методом, а именно: одностадийность, отсутствие взрывоопасных промежуточных веществ, высокая безопасность и экологичность процесса [93]. Таким образом, вместо траты средств на нейтрализацию закиси азота новый процесс использует её как ценное химическое сырье. По результатам пилотных испытаний фирмой принято решение о строительстве крупного промышленного завода мощностью 140 тыс. тонн фенола в год.
Как показывают оценки, новый процесс будет выгодным и в случае специального производства закиси азота, которая может быть с высокой селективностью получена путем окисления аммиака молекулярным кислородом [94]. С этой целью между Институтом катализа и фирмой «Solutia» заключендополнительный договор на разработку крупномасштабного процесса получения закиси азота. [95].
Таблица 2
Заключение
В работе были приведены основные синтетические методы производства фенола, их преимущества и недостатки. Наряду с ранее использовавшимися способами были изложены современные альтернативные тенденции в области улучшения технологических, экономических и экологических показателей этого процесса. Были рассмотрены научные изыскания по данной тематике. Особое место в данной работе уделяется получению фенола методом окисления бензола закисью азота, как наиболее перспективному и современному методу, упрощающему не только технологический процесс, но и решающему экологическую проблему утилизации закиси азота.
Список литературы
1. Кружалов Б.Д., Голованенко Б.И.
Совместное получение фенола и ацетона. М.: Наука, 1963, 200 с.
2. Харлампович, Г.Д. и Чркин Ю. В.
Фенолы. М.: «Химия», 1974, 376 с.
3. Юкельсон И.И.
Технология основного органического синтеза М.: «Химия», 1968, 848 с.
4. Kirk-Othmer.
Encyclopedia of Chemical Technology. 3rd Edition, v.17, p. 373, see article «Phenol».
5. Brownstain A.M.
CHEMTECH, September, 1994, p. 58.
6. Miki J., Asanuma M., Tashibana Y., Shikoda T.
Bull. Chem. Soc. Japn., 1995, v. 68, p. 2429.
7. ChemExpo, Phenol (1999, March 29) http://www.chemexpo.com/news/PROFILE990329.cfm
8. Полищук В.
Химия и жизнь, 1988, № 8, с. 68.
9. Авт. свид. СССР № 7238 и № 7240 от 7.01.1947.
10. Толстиков Г.А.
Панорама нефтехимии, 1997, № 4, с. 76.
11. Харлампович Г.Д., Чуркин Ю.В.
Фенолы. М.: Химия, 1974, 376 с.
12. SRI report No. 22C, 2-2 (Appl. Catal. A, 1994, v. 114, № 1, p. N2).
13. Марек Л.Ф., Ган Д.А.
Каталитическое окисление органических соединений. М.: ОНТИ, 1936, с. 446.
14. Голодец Г.И.
Гетерогенно-каталитическое окисление органических веществ. Киев: Науковадумка, 1978, с. 209.
15. Sheldon R.A.
Top. Curr. Chem., 1993, v. 164, p. 21.
16. Sasaki K., Kitano T., Nakai T. e. a.
New Developments in Selective Oxidation II, Elsevier, 1994, p. 451.
17. Zakharov V.Yu., Zakharova O.M., Romanovsky B. V., Mardaleishvili R.E.
React. Kinet. and Catal. Lett., 1977, v. 6, p. 133.
18. Sheldon R.A., Arends I.W.C.E., Lempers H.E.B.
Catal. Today, 1998, v. 41, p. 387.
19. Караханов Э.А., Волков С.М., Дедов А.Г.
Катализ. М.: изд. МГУ, 1987, с. 147.
20. Notary B.
Adv. Catal., 1996, v. 41, p. 253.
21. Belussi G., Rigutto M.S.
Stud. Surf. Sci. Catal., 1994, v. 85, p. 177.
22. De Vos D.E., Buskens P.L., Vanoppen D.L. e.a.
Compr. Supramol. Chem., 1996, v. 7, p. 647.
23. Arends I.W.C.E., Sheldon R.A., Wallau M., Schuchardt U.
Angew. Chem., 1997, Bd. 36, S. 1144.
24. Заявка 62-67038, Япония. Заявл. 20.09.85, № 60-206234, опубл. 26.03.87. МКИ С 07 С 37/60, С 07 С 39/04.
25. Sittig M.
Organic Chemical Process Encyclopedia. 2nd ed., 1969, p. 517.
26. Mori M., Nakai T., Jahiro H., Nata M., Sasaki K.
Bull. Chem. Soc. Jap., 1995, v. 68, № 6, p. 1747.
27. Shengchun CaO e. a.
Chin. J. Petrochem. Tech., 1995, v. 10, p. 708 [Chem. Abstracts, 1995, v. 123].
28. Moro-oka Y., Akita M.
Catal. Today, 1998, v. 41, p. 327.
29. Kuznetsova N.I., Detusheva L.G., Kuznetsova L.I. e.a.
J. Mol. Catal. A: Chemical, 1996, v. 114, p. 131.
30. Шилов Н.А.
О сопряженных реакциях окисления. М., 1905, 304 с.; Нагиев Т.М.
Химическое сопряжение. М.: Наука, 1989, 216 с.
31. Clerici M. G., Ingallina P.
Catal. Today, 1998, v. 41, p. 351.
32. Kitano T., Wani T., Ohnishi T. e.a.
Catal. Lett., 1991, v. 11, p. 11.
33. Jintoku T., Nishimura K., Takaki K., Fujiwara Y.
Chem. Lett., 1991, v. 193.
34.Иоффе
И
.
И
.,
Левин
Я
.
С
.,
Кронич
И
.
Г
.
Ж. физ. химии, 1959, т. 33, № 4, с. 863.
35. Sasaki K., Ito S., Sahek Y., Kinoshita T., Yamasaki T., Harada J.
Chem. Lett., 1983, v. 37.
36. Kitajima N., Ito M., Fukui H., Moro-oka Y.
J. Chem. Soc. Chem. Communs., 1991, p. 102.
37. Vayenas C.G., Bebelis S.I.
Stud. Surf. Sci. Catal., 1997, v. 110, p. 77.
38. Galvita V.V., Belyaev V.D., Demin A.K., Sobyanin V.A.
Appl. Catal. A: General, 1997, v. 165, p. 301.
39. Otsuka K., Yamanaka I.
Catal. Today, 1998, v. 41, p. 311.
40. Iwamoto M., Matsukami K., Kagawa S.
J. Phys. Chem., 1983, v. 87, p. 903.
41. Chem. Econ. Eng. Rev., 1982, v. 14, p. 47.
42. Process Econ. Intern., 1983, v. 4, p. 48.
43. Харитонов А.С., Соболев В.И., Панов Г.И.
Успехи химии, 1992, т. 61, № 11, с. 2062.
44. Suzuki E., Makashiro K., Ono Y.
Chem. Soc. Jap. Chem. Communs., 1988, p. 953.
45. Gubelmann M.H., Tirel P.J.
Pat. 2 630 735 (Fr.).
46. Харитонов А. С., Александрова Т. Н., Вострикова Л. А., Соболев В. И., Ионе К. Г., Панов Г. И.
А.с. СССР № 1805127 (Заявка № 4445646 от 22.06.88).
47. Burch R., Howitt C.
Appl. Catal. A: General, 1992, v. 86, № 2, p. 139.
48. Zholobenko V.
Mendeleev Communs., 1993, v. 1, p. 28.
49. Yoo J.S., Sohail A.R., Grimmer S.S., Chin C.-F.
Catal. Lett., 1994, v. 29, p. 299.
50. Bogdan V.I., Kustov L.M., Batizat D.B. e.a.
Stud. Surf. Sci. Catal., 1995, v. 94.
51. H
д
fele N., Reitzmann A., Roppelt D., Emig G.
Appl. Catal. A: General, 1997, v. 150, p. 153.
52. Kowalak S., Nowinska K., Swiecicka M. e.a.
Proc.: 12th Int. Zeolite Conf., 1998, Baltimore, MRS, v. 4, p. 2847.
53. Nowinska K., Kowalak S., Sopa M., Swiecicka M.
Proc.: 3rd Polish-German Zeolite Coll., 1998, Torin: Nicholas Copernicus University Press, p. 215.
54. Motz J.L., Heinichen H., H
ц
lderich W.F.
J. Mol. Catal. A: Chemical, 1998, v. 136, p. 175.
55. Vereshchagin S.N., Kirik N.P., Shishkina N.N., Anshits A.G.
Catal. Lett., 1998, v. 56, p. 145.
56. Ushiroguchi T., Sugimoto T.
Jap. Patent 10-25260, Jan. 27, 1998.
57. Kuhnle A., Duda M.
Eur. Pat. Appl. (1999), EP 953558 A1 19991103.
58. Thayer A.M.
Chem. Eng. News, 1998, April 6, p. 21.
59. Sobolev V.I., Dubkov K.A., Paukshtis E.A. e.a.
Appl. Catal. A, 1996, v. 141, p. 185.
60. Pirutko L.V., Ivanov D.P., Dubkov K.A. e.a.
Proc.: 12th Int. Zeolite Conf., 1998, Baltimore, p. 1245.
61. Panov G.I., Sheveleva G.A., Kharitonov A.S. e.a.
Appl. Catal. A, 1992, v. 82, p. 31.
62. Шевелева Г.А., Харитонов А.С., Панов Г.И. и др.
Нефтехимия, 1993, № 6, с. 530.
63. Sobolev V.I., Panov G.I., Kharitonov A.S.
e
.
a
.
J. Catal., 1993, v. 139, p. 435.
64.Filatov
M
.
J
.,
Pelmenschikov
A
.
G
.,
Zhidomirov
G
.
M
.
J. Mol. Catal., 1993, v. 80, p. 243.
65. Yakovlev A.L., Zhidomirov G.M.
Ibid., 1999, v. 91, p. 91.
66. Knops-Gerits P.P., Labinger J.A., Davis M.E.
Proc.: Europacat-4 ISO Workshop, 1999, Rimini, p. 129.
67. Panov G.I., Sobolev V.I., Kharitonov A.S.
J. Mol. Catal., 1990, v. 61, p. 85.
68. Дубков К.А., Соболев В.И., Панов Г.И.
Кинетика и катализ, 1997, т. 38, № 6, с. 1; Харитонов А.С., Александрова Т.Н., Панов Г.И. и др.
Кинетика и катализ, 1994, т. 35, № 2,
с. 296.
69.Panov
G
.
I
.,
Uriarte
A
.
K
.,
Rodkin
M
.
A
.,
Sobolev
V
.
I
.
Catal. Today, 1998, v. 41, p. 365.
70.Zhidomirov
G
.
M
.,
Yakovlev
A
.
L
.,
Kachurovskaya
N
.
A
.,
Yudanov
I
.
V
.
Ibid., 1999, v. 51, p. 397.
71. Боресков Г.К.
Гетерогенный катализ. М.: Наука, 1986, 303 с.
72. Боресков Г.К., Музыкантов В.С., Панов Г.И., Поповский В.В.
Кинетика и катализ, 1969, т. 10, № 5, с. 1043.
73. Che M., Tench A.J.
Adv. Catal., 1983, v. 32, p. 1.
74. Шилов А.Е., Шульпин Г.Б.
Активация и каталитические реакции углеводородов. М.: Наука, 1995, 327 с.
75.Panov
G
.
I
.,
Sobolev
V
.
I
.,
Dubkov
K
.
A
.,
Kharitonov
A
.
S
.
Stud. Surf. Sci. Catal., 1996, v. 101, p. 493.
76. Dubkov K.A., Sobolev V.I., Talsi E.P. e.a.
J. Mol. Catal., 1997, v. 123, p. 155.
77. Ионе К.Г.
Полифункциональный катализ на цеолитах. Новосибирск: Наука, 1982. 230 с.; Ratnasami
P
.,
Cumar
R
.
Catal. Today, 1991, v. 9, p. 328.
78. Pirutko L.V., Parenago O.O., Lunina E.V. e.a.
React. Kinet. and Catal. Lett., 1994, v. 52, № 2, p. 275.
79. Panov G.I., Kharitonov A.S., Fenelonov V.B. e.a.
Zeolites, 1995, v. 15, № 3, p. 253.
80. Pirutko L.V., Dubkov K.A., Solovyeva L.P., Panov G.I.
React. Kinet. and Catal. Lett., 1996, v. 58, № 1, p. 105.110.
81.Пирютко
Л
.
В
.,
Харитонов
А
.
С
.,
Панов
Г
.
И
.,
Бухтияров
В
.
И
.
Кинетикаикатализ, 1997, т. 38, № 1, с. 102.
82. Panov G.I., Sobolev V.I., Dubkov K.A. e.a.
React. Kinet. and Catal. Lett., 1997, v. 61, No. 2, p. 251.
83. Ованесян Н.С., Соболев В.И., Дубков К.А. и др.
Изв. Акад. наук, Сер. хим., 1996, т. 6, с. 1583.
84. Ованесян Н.С., Штейнман А.А., Соболев В.И. и др.
Кинетика и катализ, 1998, т. 39, № 6, с. 863.
85. Filatov M., Elizarova G., Gerasimov O. e.a.
J. Mol. Catal., 1994, v. 91, p. 71.
86. Feng X., Hall W.K.
J. Catal., 1997, v. 166, p. 368.
87. Voskoboinikov T.V., Chen H.-Y., Sachtler W.M.H.
Appl. Catal. B: Environmental, 1998, v. 19, p. 279.
88. Lazar K., Lejeune G., Ahedi R.K., Shevade S.S., Kotasthane A.N.
J. Phys. Chem. B, 1998, v.102, p. 4865.
89. Lobree L.J., Hwang I.-C., Reimer J.A., Bell A.T.
J. Catal., 1999, v. 186, p. 242.
90. Joyner R., Stockenhuber M.
J. Phys. Chem. B, 1999, v. 103, p. 5963.
91. Bordiga S., Buzzoni R., Geobaldo F. e.a.
J. Catal., 1996, v. 158, p. 486.
92. Delmon B.
Appl. Catal. B: Environmental, 1992, v. 1, p. 139.
93. Uriarte A.K., Rodkin M.A., Gross M.J. e.a.
Stud. Surf. Sci. Catal., 1997, v. 110, p. 857.
94. Патент РФ № 2102135, 1998.
95.Г. И. Панов, А. С. Харитонов Прогресс в области окислительного катализа: окисление бензола в фенол закисью азотаУДК 547.532:542.943
|
