ПЛАН:
1. Теоретические основы кинетического метода анализа.
2. Каталитические методы анализа.
3. Основные методы обработки кинетических данных.
4. Основные приемы кинетических методов анализа
5. Применение кинетических методов анализа в аналитическом контроле.
6. Список использованной литературы.
1.ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МЕТОДА
Скорость химических реакций имеет большое значение в аналитической химии. Измерение скорости реакций лежит в основе кинетических методов анализа. Медленное протекание некоторых реакций затрудняет анализ. Иногда, наоборот, небольшая скорость химических процессов благоприятствует выполнению анализа и играет положительную роль. Исследование скорости реакций – предмет химической кинетики.
Скорость реакций определяется количеством веществ, прореагировавших за единицу времени. Рассмотрим реакцию между веществами А и В:
A + B = C + D.
Ее скорость зависит от концентрации реагирующих компонентов:
V = dx/dt = K [A] [B], (1)
где х – количество вступившего в реакцию [A] (или [B] ) за время t; К – константа скорости реакции.
В самом деле, при увеличении концентрации возрастает общее количество частиц в единице объема, число их столкновений между собой и, следовательно, вероятность взаимодействия; поэтому за единицу времени прореагирует больше вещества, чем при малых концентрациях.
Физический смысл константы скорости выясняется, если принять концентрации [A] и[B] равными 1 моль/л. Тогда
V = dx/dt = K, (2)
т.е. константа скорости равна количеству вещества, прореагирующего за единицу времени при [A] = [B] = 1 моль.
Скорость реакций может изменяться в очень широких пределах – от малых долей секунды до нескольких часов или дней. Условно все реакции обычно делят на быстрые и медленные. К быстрым реакциям относят такие, в которых половина имеющегося количества вещества реагирует за 10 с или за меньшее время, все остальные реакции относят к медленным.
Очень быстрые и очень медленные реакции в химико- аналитических целях являются малопригодными. Хотя трудно установить какие-либо жесткие правила или критерии, ограничивающие применение тех или иных реакций в кинетических методах анализа, все же некоторые пределы применимости можно отметить. Почти повсеместно принято считать, что аналитическая реакция ( в кинетических методах ее часто называют индикаторной реакцией) должна продолжаться не менее 1 мин и не более 2 ч. Более быстрые реакции, как правило, не применяются потому, что при использовании обычного обычного оборудования химико –аналитической лаборатории скорость таких реакций трудно измерить с достаточной точностью. При наличии специального оборудования эти затруднения отпадают и соответствующее ограничение снимается. Реакции, продолжающиеся более 2 ч, нежелательны из-за большой длительности анализа. Оптимальным временем для измерения скорости реакции считается 10 – 15 мин. Указанные пределы в значительной степени условны, поскольку можно в достаточно широких пределах регулировать скорость химической реакции, например, изменением температуры, концентрации реагирующих веществ или введением в раствор катализаторов ( или ингибиторов).
Реклама

Примером быстрых реакций могут служить многие реакции между ионами противоположного знака заряда, например реакции между водородными и гидроксильными ионами, между окислителями и восстановителями и т.д. Однако существует и очень медленные реакции. Так, исходя из значения стандартных потенциалов, следовало ожидать, что должна происходить реакция между перманганатом и водой с выделением кислорода. Но эта реакция происходит с очень малой скоростью, в связи с чем водные растворы перманганата калия вполне устойчивы и не изменяют своей концентрации длительное время.
Известны и другие реакции, характеризующиеся большими значениями констант равновесия, но протекающие с очень малой скоростью. Наоборот, реакции с меньшими константами часто протекают быстро. Примером могут служить следующие реакции:
I2
+ AsO2
-
+ 2H2
O = 2I-
+ AsO4
3-
+ 4H+
, (3)
H2
O2
+ AsO2
-
= AsO4
3-
+ 2H+
. (4)
Реакция (3) – быстрая, а реакция (4) – очень медленная. В то же время окислительный потенциал иода значительно меньше, чем у пероксида водорода, и, следовательно, константа равновесия реакции (3) меньше, чем реакции (4). Значения констант равновесия можно найти по уравнению:
lg K = (E01
– E02
)n / 0,058. (5)
Имея в виду, что Е0
( I2
/2I-
) = 0,62 B, E0
(H2
O2
/H2
O) = 1,77 B и E0
(AsO4
3-
/AsO2
-
) = 0,56 В, для реакций (3) и (4) получаем соответственно:
lg K3
= 2,1 lg K4
= 41,7,
т.е. в термодинамическом смысле реакция (4) значительно более вероятна, чем реакция (3), и должна быть сдвинута в правую сторону в большей степени, однако скорости обеих реакций находятся в обратно пропорциональном отношении к значениям констант равновесия.
Реклама

Пользуясь различными скоростями реакций, нередко удается определять одно вещество в присутствии другого даже в том случае, если реагент взаимодействует с обоими веществами. Рассмотрим две реакции:
2Ce(SO4
)2
+ HAsO2
+ 2H2
O = Ce2
(SO4
)3
+ H3
AsO4
+ H2
SO4
, (6)
2Ce(SO4
)2
+ 2FeSO4
= Ce2
(SO4
)3
+ Fe2
(SO4
)3
(7)
сульфат церия окисляет и мышьяк, и железо (II). Однако реакция (6) идет с очень малой скоростью, в то время как реакция (7) происходит мгновенно. Таким способом удается оттитровать железо в присутствии мышьяка. Затем прибавляют катализатор OsO4
, который ускоряет реакцию (6), и оттитровывают мышьяк.
Другой пример – титрование иода тиосульфатом в растворе, содержащем свободную кислоту. Тиосульфат взаимодействует и с иодом, и с кислотой:
I2
+ 2Na2
S2
O3
= 2NaI + Na2
S4
O6
, (8)
2HCl + Na2
S2
O3
= H2
SO3
+ 2NaCl. (9)
правильные результаты определения иода по реакции (8) можно получить только потому, что скорости обеих реакций различаются во много раз. Реакция (9) протекает настолько медленно, что в процессе восстановления иода тиосульфат не успевает разложиться в соответствии с уравнением (9).
Скорость реакции можно регулировать различными способами. Из уравнения (1) видно, что скорость зависит от концентрации реагирующих веществ; следовательно, быстрая реакция замедляется при уменьшении концентрации А и В, и, наоборот, она возрастает, если концентрации соответствующих веществ увеличиваются.
Скорость реакций зависит от температуры. Зависимость константы скорости реакции от температуры определяется уравнением Аррениуса
d ln K/dT = E/RT2
, (10)
где К – константа скорости; Е – энергия активации.
В случае простых реакций, идущих в одну стадию, параметр Е показывает, какой минимальной энергией ( в расчете на 1 моль) должны обладать реагирующие частицы, чтобы они могли вступить в химическую реакцию. Частицы, энергия которых больше или равна Е, называют активными.
В среднем повышение температуры на 100
С приводит к увеличению скорости реакций в растворе приблизительно в 2-3 раза. Этот прием часто используют в анализе. Так, реакция меду щавелевой кислотой и перманганатом в холодных растворах идет с очень маленькой скоростью, однако нагревание до 80 – 900
С значительно ускоряет реакцию. Растворение металлов или их солей идет значительно быстрее при нагревании. При осаждении малорастворимых соединений нагревание раствора способствует увеличению скорости движения ионов в растворе и приводит к быстрому росту центров кристаллизации и вследствие этого к образованию крупнокристаллических осадков. В кинетических и каталитических методах анализа нередко необходимо в определенный момент времени замедлить или вообще остановить реакцию – охлаждение раствора является одним из методов такого замедления.
На скорость реакций влияет характер растворителя. Имеет значение диэлектрическая проницаемость растворителя. Для наиболее распространенного в аналитической химии случая, когда реагируют между собой ионы противоположного знака, скорость реакции уменьшается с увеличением диэлектрической проницаемости. Большинство органических растворителей имеет диэлектрические проницаемости меньше, чем у воды, и поэтому скорость реакций в таких растворителях больше, чем в водных растворах. Однако такая корреляция наблюдается далеко не всегда. Она справедлива только в пределах группы растворителей одного гомологического ряда или для серии смесей переменного состава, приготовленных из определенной пары растворителей. Существенное влияние на скорость химических реакций имеет сольватация реагентов и активированного комплекса. Сольватация последнего понижает его энергию, что в конечном счете ускоряет химическую реакцию. Растворители по их сольватирующей способности можно классифицировать следующим образом: 1) протонные растворители – легко отщепляют протон и обычно содержат группы –ОН, =NH, их отличительная особенность – способность к образованию водородной связи. Типичные растворители этого типа: вода, спирты, карбоновые кислоты, фенолы, аммиак; 2) апротонные растворители - не имеют кислотного водорода, они хорошо сольватируют катионы, практически не сольватируя анионы. Типичные растворители этого типа : ацетон, сульфолан, диметилформамид, диэтиловый эфир, диоксан; 3) инертные растворители характеризуются малой диэлектрической проницаемостью и малым дипольным моментом. К таким растворителям относятся алканы, алкены, ароматические углеводороды, четыреххлористый углерод, сероуглерод. Эти растворители обладают малой сольватирующей активностью, ускоряя химические реакции в основном за счет гомогенизации реакционной среды; 4) растворители с электрофильными свойствами представлены сильными кислотами или суперкислотными системами, например, FSO3
H, CF3
COOH, FSO3
H/SbF5
, кислоты Льюиса.
Скорость реакций зависит от ионной силы раствора. В наиболее простом случае, когда взаимодействуют ионы А и В в растворе, зависимость логарифма константы скорости К от ионной силы раствора выражается следующим уравнением:
lg K = lg K0
+ aZA
ZB
√μ, (11)
где K0
– константа скорости реакции в среде, для которой коэффициенты активности исходных частиц А и В и активированного комплекса между ними приняты равными единице; а – константа, включающая величины ддиэлектрической проницаемости растворителя и абсолютной температуры; Za
и ZB
– заряды частиц; μ – ионная сила раствора.
Из уравнения видно, что с увеличением ионной силы, т.е. с введением в реакционную систему посторонних хорошо диссоциирующих солей, скорость реакции между ионами противоположного знака заряда уменьшается. Объяснение заключается в том, что ионы посторонних солей образуют вокруг реагирующих ионов ионную атмосферу из ионов противоположного знака заряда, которая препятствует непосредственному контакту между ионами и на разрушение которой необходимо затратить определенное время. Наоборот, если реагируют между собой ионы с зарядами одинакового знака, скорость реакции должна возрастать.
Изменение концентрации ионов водорода в рекциях окисления – воостановления влияет не только на окислительный потенциал системы, в соответствии с уравнением Нернста, но также нередко на скорость реакции. Так, окислительные потенциалы систем Fe3+
/Fe2+
и I2
/2I-
не изменяются в зависимости от рН, однако скорость реакции
2Fe3+
+ 2I-
= 2Fe2+
+I2
(12)
сильно возрастает с увеличением концентрации водородных ионов раствора. Аналогичное влияние на скорость процесса оказывает рН раствора реакции
2Cu2+
+ 4I-
= Cu2
I2
+ I2
(13)
использующейся для иодометрического определения меди. Видно, что ионы водорода не участвуют в суммарном уравнении реакции и поэтому не влияют на окислительный потенциал реагирующих веществ. Однако при слишком малой кислотности раствора реакция (13) проходит очень медленно и ее скорость возрастает с уменьшением рН раствора.
Скорость реакций зависит от введения катализаторов. Катализаторы могут ускорять или замедлять реакции, в последнем случае говорят об ингибирующем действии. Ускорение реакций связано с тем, что катализатор, взаимодействуя с реагентами, образует неустойчивые промежуточные активные соединения (одно или несколько), понижая энергию активированных комплексов (одного или нескольких). Природа такого взаимодействия зависит от типа катализа. В реакциях имеют место гомогенный, гетерогенный, ферментативный катализы. Методы, основанные на каталитическом действии вещества, представляют собой разновидность кинетических методов, их называют каталитическими.
2. КАТАЛИТИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Каталитические методы анализа – вариант кинетических методов. Они основаны на измерении скорости химической реакции, протекающей в растворе при действии катализатора; нередко катализатор является определяемым веществом. Известно много медленных реакций, скорость которых в определенных условиях увеличивается пропорционально концентрации введенного катализатора по концентрации введенного катализатора. Это дает возможность определить количество катализатора по концентрации продуктов реакции, образующихся за определенный промежуток времени.
Для определения различных неорганических веществ используется несколько десятков каталитических реакций следующих типов: 1) окислительно-восстановительные реакции (окисление перекисью водорода, галогенатами, солями церия (IV) и др.). 2) реакции изотопного обмена между одноименно заряженными ионами (Се4+
- Се3+
, АuCl-
4
– Cl-
и др.). 3) реакции, приводящие к возникновению каталитических полярографических токов. 4) реакции замещения во внутренней сфере комплексных соединений (например, замещение CN-
в Fe(CN)4-
6
водой. 5) Гетерогенно-каталитические реакции.
Наиболее распространены каталитические методы с использованием реакций окисления-восстановления.
Окислительно–восстановительные реакции катализируются преимущественно ионами переходных металлов, характеризующихся большой склонностью к образованию комплексных соединений и способностью изменять степень окисления в растворе. Указанные две особенности имеют существенное значение для объяснения каталитического действия. Рассмотрим, например, медленную реакцию между окислителем Ок1
и восстановителем В2
:
Ок1
+ В2
↔ В1
+ Ок2
, (14)
где индексы 1 и 2 обозначают первое и второе вещество в окисленной и восстановленной формах соответственно.
Действие катализатора К обусловлено тем, что его окисленная форма КОк
быстро реагирует с восстановителем В2
, а образовавшаяся восстановленная форма КВ
также быстро переходит в окисленную форму КОк
при действии окислителя Ок1
. Таким образом, механизм катализа можно схематически выразить следующими уравнениями:
КОк
+ В2
↔ КВ
+ Ок2
( быстрая реакция), (15)
КВ
+ Ок1
↔ КОк
+ В1
( быстрая реакция). (16)
Суммирование этих уравнений приводит к уравнению (14), причем после завершения цикла катализатор снова находится в своей первоначальной форме. Механизм катализа может быть также связан с образованием нестойких промежуточных комплексных соединений катализатора с веществами Ок1
или В2
. Эти комплексы реагируют с основными участвующими в реакции веществами Ок1
и В2
значительно быстрее, чем эти вещества взаимодействуют между собой.
Выяснение механизма каталитических реакций является сложной задачей и требует в каждом отдельном случае проведения специальных исследований.
Главная особенность каталитических методов определения – их высокая чувствительность. Это обусловлено тем, что одна и та же частица катализатора действует многократно, вовлекая в реакцию большие количества веществ Ок1
и В2
, и за короткий период времени способствует образованию продуктов реакции, концентрация которых во много раз больше концентрации введенного катализатора.
Для определения органических веществ и некоторых неорганических используют каталитические ферментативные методы. Ферменты – это белковые молекулы, которые катализируют химические реакции в биологических системах. Их часто называют биокатализаторами. Ферментативный катализ интересен сочетанием особенностей гомогенного и гетерогенного катализов.
Для того чтобы обеспечить многократность использования ферментов, их подвергают иммобилизации. Иммобилизацией называют перевод ферментов, обычно растворимых в воде, в водонерастворимое состояние с сохранением их каталитической активности. Молекулу фермента присоединяют за счет образования ковалентной связи к какому-либо водонерастворимому носителю – целлюлозе, стеклу, бумаге, силикагелю, полистиролу и др. Можно также диспергировать фермент в геле какого-либо вещества.
Каталитические ферментативные методы анализа используют преимущественно для определения многих органических соединений, в частности таких, которые содержатся в биологических объектах ( в крови, моче, тканях и др.). Таким способом можно определять мочевину. Мочевую кислоту, аминокислоты и другие органические кислоты, глюкозу и другие сахара, антибиотики и т.д.
Пределы обнаружения названных и многих других органических веществ – субстратов ферментативными реакциями находятся обычно в интервале 10-4
- 10-8
М.
Определение каталитическим ферментативным методом обычно основано на измерении скорости изменения концентрации одного из участников реакции при постоянной концентрации фермента. Например, определение глюкозы основано на следующей реакции:
Глюкоза + О2
= Глюконовая кислота + Н2
О2
.
Эта реакция катализируется ферментом глюкозоксидазой. Скорость реакции определяют по количеству выделившегося пероксида водорода. Для идентификации последнего можно использовать реакцию окисления какого-либо бесцветного органического вещества, окисленная форма которого окрашена, например, окисление о-толуидина. Тогда скорость увеличения оптической плотности прямо пропорциональна исходной концентрации глюкозы; необходимо только, чтобы кислород и восстановленная форма красителя находились в большом избытке по отношению к количеству определяемой глюкозы.
Для определения количества вещества ферментативными методами применяют и другие приемы. Так, ионы многих металлов являются ингибиторами каталитического действия ряда ферментов; они снижают их каталитическую активность в определенной индикаторной реакции. Степень этого снижения при некоторых условиях пропорциональна содержанию ионов определяемого металла в анализируемом растворе. Например, ионы ртути ингибируют каталитическую активность уреазы в реакции разложения мочевины
уреаза
 NH2
CONH2
+ H2
O 2NH4
+
+ CO2
. NH2
CONH2
+ H2
O 2NH4
+
+ CO2
.
Скорость этой реакции уменьшается пропорционально концентрации ионов ртути, и этим методом можно определять ничтожные количества металлов.
Пределы обнаружения ингибиторов очень низки. Так, применяя различные ферментативные реакции, можно определить 10-11
г/мл ртути, 10-12
г/мл меди, 8 * 10-12
г/мл цинка и т.д.
3.ОСНОВНЫЕ МЕТОДЫ ОБРАБОТКИ КИНЕТИЧЕСКИХ ДАННЫХ
Как указывалось выше, в кинетичесуких методах анализа измеряемым свойством системы, на основании которого делают выводы о концентрации вещества, является скорость химической реакции. Хотя в кинетических методах анализа, в принципе, могут быть использованы любые реакции, скорость которых может быть измерена достаточно точно, все же наиболее часто применяют так называемые каталитические реакции, скорость которых зависит от концентрации катализатора. При наличии в растворе катализатора в кинетическом уравнении (1) появляется соответствующий сомножитель :
dx/ dt = K CK
[A][B], (16)
где СК
– концентрация катализатора.
Концентрацию одного из участников реакции, например, вещества В, можно взять заведомо в большом избытке, так что его убыль в результате протекания реакции будет пренебрежимо мала, и, следовательно можно записать kb = χ, тогда
dx /dt = χ CK
[A]. (17)
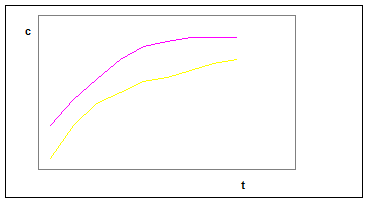
На рис. 1 приводятся типичные кинетические кривые. График показывает возрастание во времени концентрации вещества Х. Кривая 1 в начальный момент времени имеет линейный участок, т.е. в начальный момент времени угловой коэффициент кинетической кривой постоянен. На кривой 2 линейный участок даже в начальный момент времени отсутствует. Различный характер кинетических кривых вызывает разные способы их обработки.
Рис.1 . Кинетические кривые.
Рассмотрим сначала кривую 1. Уравнение (17) показывает, что величина dx/dt может быть постоянной только при условии постоянства [А], т.е. если концентрация вещества А в ходе реакции существенно меняться не будет. Это уравнение является основой различных вариантов кинетического метода, названных дифференциальными. Интегрирование дает:
х = χ СК
[А] t. (18)
Несколько сложнее обработка данных, представленных кривой 2. Здесь нет области, в которой [А] = const, поэтому приходится интегрировать кинетическое уравнение (17).Разделив переменные и проинтегрировав, получаем
∫dx /(a – x) = ∫ χ CK
dt
или
-ln (a – x) = χ CK
t + const.
Постоянную интегрирования находим из начальных условий: при t = 0, х = 0 и, следовательно, - ln а = const . Окончательно можно записать:
ln a / (a – x) = χ CK
t. (19)
Методы анализа, основанные на применении этого уравнения, называют интегральными.
4.ОСНОВНЫЕ ПРИЕМЫ КИНЕТИЧЕСКИХ МЕТОДОВ АНАЛИЗА
Уравнения (17) – (19) в явном виде связывают кинетические характеристики реакции с концентрацией катализатора. Как видно, концентрация катализатора может быть найдена или непосредственно по скорости реакции, или по времени ее протекания, или по концентрации образующихся продуктов. В зависимости от того, какое свойство или какая характеристика реакции используется для определения концентрации, выделяют методы тангенсов, фиксированного времени, фиксированной концентрации.
Известны, кроме того, и другие методы, имеющие более частный характер, например методы индукционного периода, непосредственного дифференцирования и т.д.
Метод тангенсов.
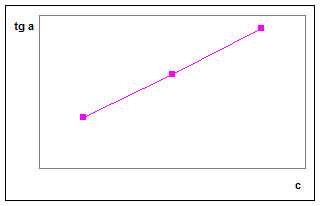
В методе тангенсов измеряют скорость реакции обычно по возрастанию концентрации одного из образующихся продуктов и строят график, аналогичный изображенному на рис.1. Если кинетическая кривая в начальный период протекания реакции имеет линейчатый характер, применяют дифференциальный вариант метода тангенсов. Уравнение ( 17) показывает, что в этом случае скорость реакции dx /dt , характеризуемая тангенсом угла наклона кинетической кривой, пропорциональна концентрации катализатора.
График в координатах тангенс угла наклона – концентрация определяемого вещества ( отсюда название «метод тангенсов») обычно линеен При анализе неизвестного раствора измеряют скорость реакции в тех же условиях, в каких она определялась для построения градуировочного графика, определяют tg α и по градуировочному графику находят концентрацию анализируемого компонента сх
.
Рис.2. Градуировочный график в методе тангенсов.
Если кинетическая кривая имеет вид кривой 2 на рис.1, т.е. линейный участок отсутствует, применяется интегральный вариант метода тангенсов. В соответствии с уравнением (19) кинетическую кривую следует строить в координатах lg a / (a – x) – t. Тангенс угла наклона прямой в этих координатах, как показывает уравнение (19) , пропорционален концентрации катализатора и, следовательно, градуировочный график будет также линеен.
Для измерения текущей концентрации очень удобны фотометрические методы, так как оптическая плотность раствора прямо пропорциональна концентрации вещества. При построении кинетической кривой на оси ординат вместо концентрации можно откладывать оптическую плотность: тангенс угла наклона для построения градуировочного графика можно вычислять как dA /dt. Метод тангенсов с успехом применяется для самых различных реакций, по точности определения он превосходит все остальные варианты кинетических методов.
Метод фиксированного времени
В методе фиксированного времени определяют концентрацию одного из участников реакции за строго определенный промежуток времени. Если, например, продукт реакции окрашен, через определенный промежуток времени измеряют оптическую плотность раствора. При небольшой глубине протекания реакции применяют дифференциальный вариант. Из уравнения (18) получаем
C = x / χat = (1/χat) x,
где t – заданный промежуток времени.
Выражение в скобках постоянно, так как а и t фиксированы. Его числовое значение может быть использовано для последующих расчетов ск
по измеренной величине х. Практически можно поступать следующим образом. Готовят серии растворов с переменной и известной концентрацией СА
( при СВ
= const), измеряют через определенный промежуток времени (t = const) количество прореагировавшего вещества х и строят градуировочный график (рис.3), откладывая на оси ординат х, а на оси абсцисс – концентрацию СА
. По этому графику легко установить затем концентрацию СА
в анализируемом растворе, поставив аналогичный эксперимент с раствором, в котором содержание СА
неизвестно. Вполне понятно, что фиксированный отрезок времени сохраняется один и тот же как при построении градуировочного графика, так и при анализе неизвестного раствора. При большой глубине протекания реакции применяется интегральный вариант, основанный на решении уравнения (19) относительно ск
. Метод фиксированного времени, как видно, проще метода тангенсов, однако по точности он ему уступает.
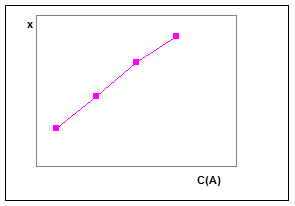 |
Рис.3. Градуировочный график в методе фиксированного времени.Метод фиксированной концентрации
В методе фиксированной концентрации измеряют время, в течение которого концентрация продукта реакции или одного из реагирующих веществ достигает определенного, заранее заданного значения. Этот метод по сути близок методу фиксированного времени. Если глубина протекания реакции невелика, используют дифференциальный вариант, так же как и в методе фиксированного времени, основаной на решении уравнения (18) относительно ск
:
Ck
= x / χat = ( x / χa) 1/t, (20)
где х – заданная концентрация продукта реакции.
Выражение в скобках постоянно, поскольку постоянны х и а, его числовое значение может быть определено по стандартному раствору. Градуировочный график в методе фиксированной концентрации, как показывает уравнение (20) , следует строить в координатах ск
- 1/t , где ск
– определяемая концентрация, а t – время, необходимое для достижения заданной концентрации продукта реакции.
При более глубоком протекании реакции испльзуется интегральный вариант, основанный на решении уравнения (19):
Ck
= (ln a / (a – x) 1/χ) 1 /t.
Градуировочный график, как видно, следует строить также в координатах ск
- 1/t. По точности метод фиксированной концентрации близок к методу фиксированного времени и уступает методу тангенсов.
Метод добавок
Этот метод обладает известными достоинствами. Сначала оапределяют скорость химической реакции в анализируемом растворе, содержащем определяемый компенент неизвестной концентрации сх
, а затем в анализируемые раствор добавляют точно известное количество определяемого компонента и вновь измеряют скорость реакции.
Метод каталиметрического титрования
Сущность этого метода можно пояснить на примере церий-арсенитной реакции, которая катализируется осмием, рутением и иодидом:
2Ce(SO4
)2
+ HAsO2
+ 2H2
O = Ce2
(SO4
)3
+ H3
AsO4
+ H2
SO4
Так как иодид является катализатором реакции церия с арсенитом, скорость этой реакции будет возрастать с увеличением концентрации иодида. Ионы Аg+
обладают ингибиторным эффектом, блокируя катализатор. При введении в раствор ионов Аg+
происходит образованиеAgI , и концентрация I-
падает, вызывая соответствующее уменьшение скорости церий-арсенитной реакции. При каталиметрическом титровании к анализируемому раствору, содержащему иодид -ион I-
в церий-арсенитной смеси, несколькими порциями добавляют титрованный раствор AgNO3
и определяют скорость реакции. Точку эквивалентности находят из графика в координатах: объем раствора AgNО3
– скорость реакции, характеризуемая угловым коэффициентом dx/dt. Этим методом можно оттитровывать иодтд в очень разбавленных растворах ( 10-6
моль/л и меньше).
5. ПРИМЕНЕНИЕ КИНЕТИЧЕСКОГО МЕТОДА АНАЛИЗА В АНАЛИТИЧЕСКОМ КОНТРОЛЕ
Кинетические методы анализа характеризуются высокой точностью и поэтому часто используются при определении малых и ультрамалых содержаний ( до 10-8
– 10-6
мкг) . Особенно эффективным оказалось применение кинетических методов для определения микропримесей в чистых и сверхчистых веществах и материалах.
В настоящее время известны десятки каталитических реакций, с помощью которых могут быть определены свыше 40 элементов периодической системы. Характеристика некоторых из них приведена в таблице 1.
Таблица 1.
| Элементы |
Чувствительность
Минимальная концентрация открываемый минимум, мкг
мкг/ мл
|
| Ванадий |
10-4
|
- |
| Вольфрам |
10-3
|
- |
| Железо |
- |
10-4
|
| Золото |
- |
10-5
|
| Иод |
- |
10-5
|
| Кобальт |
- |
10-6
|
| Магний |
- |
100 |
| Марганец |
- |
10-5
|
| Медь |
10-4
|
- |
| Молибден |
10-4
|
- |
| Ниобий |
- |
0,5 |
| Осмий |
- |
10-3
|
| Палладий |
10-2
|
- |
| Платина |
10-2
|
- |
| Рений |
- |
10-3
|
| Ртуть |
- |
10-2
|
| Рутений |
- |
10-3
|
| Свинец |
- |
10-2
|
| Селен |
- |
10-5
|
| Сера |
- |
10-4
|
| Серебро |
- |
10-5
|
| Тантал |
10-2
|
- |
| Теллур |
- |
10-2
|
| Титан |
- |
0,5 |
| Торий |
0,1 |
- |
| Уран |
10-3
|
- |
| Фтор |
2 |
- |
| Хром |
10-3
|
- |
| Цирконий |
1 |
- |
Например, реакция иодида с пероксидом водорода без катализатора идет очень медленно:
H2
O2
+ 2H+
+ 2I-
= I2
+ 2H2
O
В присутствии следов молибдена, вольфрама, циркония, гафния, ниобия, тантала и других элементов она проходит за несколько минут. Скорость реакции легко определяется по возрастанию оптической плотности иодкрахмального раствора в единицу времени. Чувствительность реакции достаточно высока ( см. табл. 1).
Интересны каталитические эффекты в реакции (6) взаимодействия мышьяковистой кислоты с солями церия (IV), которая катализируется осмием, рутением и иодидом. Исследование показало. Что скорость этой реакции, катализируемой осмием, не зависит от концентрации церия, но увеличивается с ростом концентрации мышьяковистой кислоты. Если в качестве катализатора выступает рутений, скорость реакции (6) перестает зависеть от концентрации мышьяковистой кислоты, но увеличивается с концентрацией церия.Эти возможности церий-арсенитной реакции открывают возможность определения осмия и рутения в одном растворе без предварительного разделения. По скорости реакции в растворах с увеличивающейся концентрацией церия можно определить рутений, а по скорости реакции при возрастающей концентрации мышьяковистой кислоты найти содержание осмия.
С помощью кинетических методов анализа в большинстве случаев определяется не общая, а равновесная концентрация реагирующих веществ. В связи с этим кинетические методы успешно применяются для изучения различных равновесий в растворах (комплексообразование, кислотно-основное взаимодействие и др.).
Наиболее ценной особенностью кинетических методов является возможность определения элементов при их содержании 10-8
– 10-6
мкг, что превосходит соответсвующую характеристику спектрального, спектрофотометрического, потенциометрического и многих других методов анализа. Анализ с помощью кинетических методов выполняется быстро и просто, без применения сложных приборов. Кинетическим методом можно определить свыше 40 элементов периодической системы с погрешностью, не превышающей ± 10 % (отн.). В некоторых случаях кинетические методы обладают достаточной специфичностью, однако, как правило, их специфичность невысока. Специфичность каталитических реакций повышают путем маскировки каталитически активных примесей при введении в анализируемый раствор соответствующих реагентов.
Кинетичекий метод можно применять для определения двух компонентов в смеси, в частности двух органических веществ, например, двух органических пероксидов. Определение возможно только в том случае, когда константы скорости реакций обоих веществ с реагентами значительно различаются. Исследуют скорость взаимодействия обоих пероксидов, например, с раствором иодида калия; о скорости реакции судят по количеству выделившегося иода. График зависимости концентрации иода ( обнаруживают посредством крахмала) от времени протекания реакции состоит из двух участков с различным наклоном. Измерив тангенсы углов наклона α1
и α2
, можно найти содержание каждого компонента, пользуясь тем, что тангенсы углов наклона пропорциональны содержанию каждого вещества.
Применяя специальные методы обработки результатов, находят содержание двух компонентов смеси и при близких скоростях обеих реакций.
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. Алесковский В.Б., Бардин В.В., Бойчинова Е.С и др. Физико-химические методы анализа. Практическое руководство. – Л.: Химия, 1988.
2. Аналитическая химия. Физические и физико-химические методы анализа / Под ред. О.М. Петрухина. – М.: Химия, 2001.
3. Васильев В.П. Аналитическая химия. Книга 2.- Физико-химические методы анализа. – М.: Дрофа, 2002.
4. Лурье Ю.Ю. Справочник по аналитической химии. – М.: Химия, 1989
Основы аналитической химии / Под ред. академика Ю.А. Золотова. - М.: Высшая школа, 2002. Кн. 1, 2.
5. Пилипенко А.Т., Пятницкий И.В. Аналитическая химия. – М.: Химия, 1990. Т. 1, 2.
6. Юинг Д. Инструментальные методы анализа. – М.: Мир, 1989.
|