|
Элементарные стадии органических реакций, катализируемых кислотами, основаниями, нуклеофильными катализаторами, комплексами металлов, твердыми металлами и их соединениями в газофазных или жидкофазных гетерогенных и гомогенных процессах, – это реакции образования и превращения различных органических и металлоорганических интермедиатов, а также комплексов металлов. К органическим промежуточным соединениям относятся ионы карбения R+
, карбония RH2
+
, карбо-анионы R–
, анион- и катионрадикалы 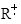 , , 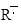 , радикалы и бирадикалы R·, R:, а также молекулярные комплексы органических донорных и акцепторных молекул (D®A), которые называют также комплексами с переносом заряда. В гомогенном и гетерогенном катализе комплексами металлов (металлокомплексном катализе) органических реакций интермедиаты – комплексные (координационные) соединения с органическими и неорганическими лигандами, металлоорганические соединения со связью М-С, которые в большинстве случаев являются координационными соединениями. Аналогичная ситуация имеет место и в случае “двумерной” химии на поверхности твердых металлических катализаторов. Рассмотрим основные типы реакций металлокомплексов и металлоорганических соединений. , радикалы и бирадикалы R·, R:, а также молекулярные комплексы органических донорных и акцепторных молекул (D®A), которые называют также комплексами с переносом заряда. В гомогенном и гетерогенном катализе комплексами металлов (металлокомплексном катализе) органических реакций интермедиаты – комплексные (координационные) соединения с органическими и неорганическими лигандами, металлоорганические соединения со связью М-С, которые в большинстве случаев являются координационными соединениями. Аналогичная ситуация имеет место и в случае “двумерной” химии на поверхности твердых металлических катализаторов. Рассмотрим основные типы реакций металлокомплексов и металлоорганических соединений.
Элементарные стадии с участием комплексов металлов
Реакции металлокомплексов можно разделить на три группы:
а) реакции переноса электрона;
б) реакции замещения лигандов;
в) реакции координированных лигандов.
Реакции переноса электронов
Два механизма реализуются в реакциях переноса электронов – внешнесферный механизм (без изменений в координационных сферах донора и акцептора) и мостиковый (внутрисферный) механизм, приводящий к изменениям в координационной сфере металла.
Рассмотрим внешнесферный механизм на примере октаэдрических комплексов переходных металлов. В случае симметричных реакций (DG
0
= 0)

константы скорости меняются в очень широком интервале значений – от 10–12
до 105
л·моль–1
·сек–1
, в зависимости от электронной конфигурации иона и степени ее перестройки в ходе процесса. В этих реакциях очень наглядно проявляется принцип наименьшего движения – наименьшего изменения валентной оболочки участников реакции.
В реакции переноса электрона (1) (Со*
– изотоп атома Со)
 (1) (1)
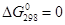 (симметричная реакция), Co2+
(d7
) переходит в Co3+
(d6
). Электронная конфигурация (валентная оболочка) в ходе этого переноса не меняется (симметричная реакция), Co2+
(d7
) переходит в Co3+
(d6
). Электронная конфигурация (валентная оболочка) в ходе этого переноса не меняется

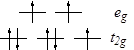
6 электронов на трижды вырожденном связывающем уровне остаются без изменения (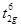 ), а с разрыхляющего eg
уровня снимается один электрон. Константа скорости второго порядка для реакции (1) k
1
= 1.1 л×моль–1
×сек–1
. Поскольку Phen (фенантролин) относится к сильным лигандам, максимальное число из 7 d
-электронов спарено (спин-спаренное состояние). В случае слабого лиганда NH3
ситуация кардинально меняется. Co(NH3
)n
2+
(n = 4, 5, 6) находится в спин-неспаренном (высокоспиновом) состоянии ), а с разрыхляющего eg
уровня снимается один электрон. Константа скорости второго порядка для реакции (1) k
1
= 1.1 л×моль–1
×сек–1
. Поскольку Phen (фенантролин) относится к сильным лигандам, максимальное число из 7 d
-электронов спарено (спин-спаренное состояние). В случае слабого лиганда NH3
ситуация кардинально меняется. Co(NH3
)n
2+
(n = 4, 5, 6) находится в спин-неспаренном (высокоспиновом) состоянии 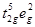 . .
Более прочный комплекс Co(NH3
)6
3+
(прочнее Co(NH3
)6
2+
~ в 1030
раз) находится в спин-спаренном состоянии 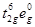 , как и комплекс с Phen. Поэтому в процессе переноса электрона , как и комплекс с Phen. Поэтому в процессе переноса электрона  должна сильно перестроиться валентная оболочка и в результате k
= 10–9
л×моль–1
×сек–1
. Степень превращения Со2+
в Со3+
, равная 50%, достигается в случае лиганда Phen за 1 секунду, а в случае NH3
~ за 30 лет. Очевидно, что стадию с такой скоростью (формально элементарную) можно исключить из набора элементарных стадий при анализе механизмов реакции. должна сильно перестроиться валентная оболочка и в результате k
= 10–9
л×моль–1
×сек–1
. Степень превращения Со2+
в Со3+
, равная 50%, достигается в случае лиганда Phen за 1 секунду, а в случае NH3
~ за 30 лет. Очевидно, что стадию с такой скоростью (формально элементарную) можно исключить из набора элементарных стадий при анализе механизмов реакции.
Реклама

Величина DG
¹
для реакции переноса электронов при образовании комплекса столкновения согласно теории Маркуса включает два компонента 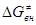 и и 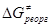
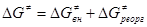 (2) (2)
Первый член – энергия реорганизации связей M-L внутри комплекса (длина и прочность связи при изменении валентного состояния). Величина 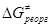 включает энергию перестройки внешней сольватной оболочки в процессе изменения координат M-L и заряда комплекса. Чем меньше изменение электронного окружения и меньше изменение длины M-L, тем ниже , чем больше по размерам лиганды, тем меньше и, в результате, выше скорость переноса электронов. Величину включает энергию перестройки внешней сольватной оболочки в процессе изменения координат M-L и заряда комплекса. Чем меньше изменение электронного окружения и меньше изменение длины M-L, тем ниже , чем больше по размерам лиганды, тем меньше и, в результате, выше скорость переноса электронов. Величину  для общего случая можно рассчитать по уравнению Маркуса для общего случая можно рассчитать по уравнению Маркуса
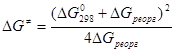 , (3) , (3)
где  . При . При 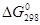 = 0 = 0 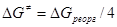 . .
В случае внутрисферного механизма процесс переноса электрона облегчается, поскольку один из лигандов первого комплекса образует мостиковый комплекс со вторым комплексом, вытесняя из него один из лигандов
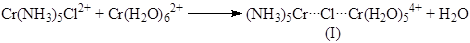
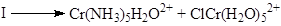
Константы скорости такого процесса на 8 порядков выше константы для восстановления Cr(NH3
)6
3+
. В таких реакциях восстанавливающий агент должен быть лабильным комплексом, а лиганд в окислителе должен быть способен к образованию мостиков (Cl–
, Br–
, I–
, N3
–
, NCS–
, bipy).
Реакции замещения лигандов
Одна из важнейших стадий в металлокомплексном катализе – взаимодействие субстрата Y с комплексом – происходит по трем механизмам:
а) Замещение лиганда растворителем. Обычно такую стадию изображают как диссоциацию комплекса
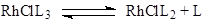 (4) (4)
Суть процесса в большинстве случаев – замещение лиганда L растворителем S, который далее легко замещается молекулой субстрата Y
 (5) (5)
б) Присоединение нового лиганда по свободной координате с образованием ассоциата с последующей диссоциацией замещаемого лиганда
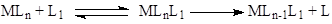 (6) (6)
в) Синхронное замещение (типа SN
2) без образования интермедиата
 (7) (7)
В случае комплексов Pt(II) очень часто скорость реакции описывается двухмаршрутным уравнением
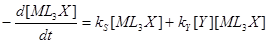 (8) (8)
где kS
и kY
– константы скорости процессов, протекающих по реакциям (5) (с растворителем) и (6) с лигандом Y. Например,
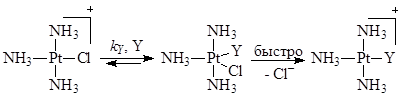
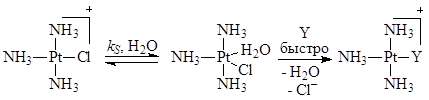
Последняя стадия второго маршрута есть сумма трех быстрых элементарных стадий – отщепления Cl–
, присоединения Y и отщепления молекулы H2
O.
В плоских квадратных комплексах переходных металлов наблюдается транс-эффект, сформулированный И.И.Черняевым – влияние LT на скорость замещения лиганда, находящегося в транс- положении к лиганду LT. Для комплексов Pt(II) транс-эффект возрастает в ряду лигандов:
Реклама

H2
O ~ NH3
< Cl–
~ Br–
< I–
~ NO2
–
~ C6
H5
–
< CH3
–
<
< PR3
~ AsR3
~ H–
< олефин ~ CO ~ CN–
.
Наличие кинетического транс-эффекта и термодинамического транс-влияния объясняет возможность синтеза инертных изомерных комплексов Pt(NH3
)2
Cl2
:
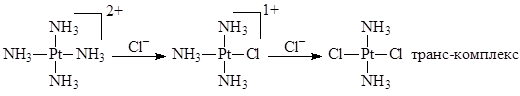
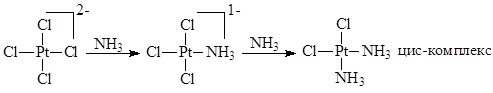
Реакции координированных лигандов
- Реакции электрофильного замещения (SE
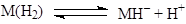
) водорода металлом в координационной сфере металла и обратные им процессы
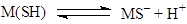
SH – H2
O, ROH, RNH2
, RSH, ArH, RCºCН.
Даже молекулы H2
и CH4
участвуют в реакциях такого типа

- Реакции внедрения L по связи M-X
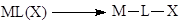
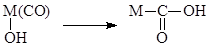
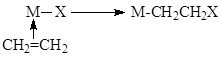
В случае X = R (металлоорганический комплекс) координированные металлом молекулы также внедряются по связи M-R (L – CO, RNC, C2
H2
, C2
H4
, N2
, CO2
, O2
и др.). Реакции внедрения есть результат внутримолекулярной атаки нуклеофила X на координированную по s- или p-типу молекулу. Обратные реакции – реакции a- и b-элиминирования
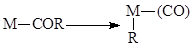 
- Реакции окислительного присоединения и восстановительного элиминирования
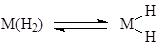
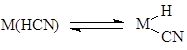
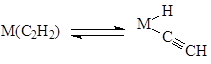
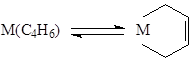
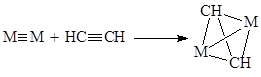
M2
(C2
H2
) º M2
4+
(C2
H2
)4–

По-видимому, в этих реакциях всегда имеет место предварительная координация присоединяемой молекулы, но это не всегда удается зафиксировать. Поэтому наличие свободного места в координационной сфере или места, связанного с растворителем, который легко замещается субстратом, является важным фактором, влияющим на реакционную способность металлокомплексов. Например, бис-p-аллильные комплексы Ni являются хорошими предшественниками каталитически активных частиц, поскольку вследствие легкого восстановительного элиминирования бис-аллила появляется комплекс [Ni0
] с растворителем, т.н. “голый” никель. Роль свободных мест иллюстрирует следующий пример:
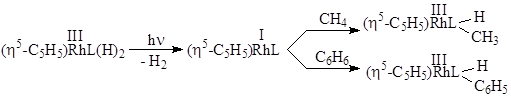
- Реакции нуклеофильного и электрофильного присоединения к p- и s-комплексам металлов


В качестве интермедиатов каталитических реакций встречаются как классические металлоорганические соединения, имеющие связи M-C, M=C и MºC, так и неклассические соединения, в которых органический лиганд координирован по h2
, h3
, h4
, h5
и h6
-типу, или является элементом электронно-дефицитных структур – мостиковые СН3
и С6
Н6
-группы, неклассические карбиды (Rh6
C(CO)16
, C(AuL)5
+
, C(AuL)6
2+
и др.).
Среди специфичных механизмов для классических s-металлоорганических соединений отметим несколько механизмов. Так, установлено 5 механизмов электрофильного замещения атома металла по связи M-C.
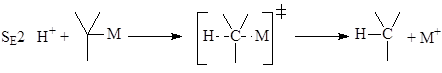
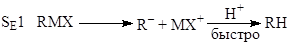
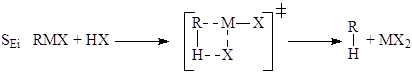
электрофильное замещение с нуклеофильным содействием
AdE Присоединение-элиминирование
AdE(C) Присоединение к атому С в sp2
-гибридизации
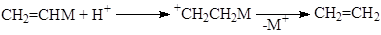
AdE(M) Присоединение окислительное к металлу
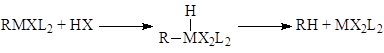
Нуклеофильное замещение у атома углерода в реакциях деметаллирования металлоорганических соединений, происходит как окислительно-восстановительный процесс:
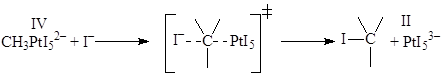
Возможно участие окислителя в такой стадии
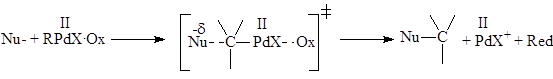
Таким окислителем может служить CuCl2
, п-бензохинон, NO3
–
и др. соединения. Приведем еще две характерные для RMX элементарные стадии:
гидрогенолиз связи M-C

и гомолиз связи M-C

Важные правилом, относящимся ко всем реакциям комплексных и металлоорганических соединений и связанным с принципом наименьшего движения, является правило 16-18-электронной оболочки Толмена (раздел 2).
Координационные и металлоорганические соединения на поверхности
Согласно современным представлениям на поверхности металлов образуются комплексы и металлоорганические соединения, аналогичные соединениям в растворах. Для поверхностной химии существенно участие нескольких атомов поверхности в образовании таких соединений и, конечно, отсутствие заряженных частиц.
Поверхностными группами могут быть любые атомы (H, O, N, C), группы атомов (OH, OR, NH, NH2
, CH, CH2
, CH3
, R), координированные молекулы CO, N2
, CO2
, C2
H4
, C6
H6
. Например, при адсорбции СО на поверхности металла обнаружены следующие структуры:
  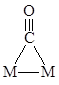 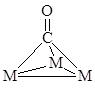 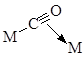
Молекула С2
Н4
на поверхности металла образует p-комплексы с одним центром и ди-s-связанные этиленовые мостики M–CH2
CH2
–M, т.е. по существу, металлоциклы
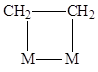 . .
На поверхности Rh, например, при адсорбции этилена, происходят следующие процессы превращения этилена по мере повышения температуры:
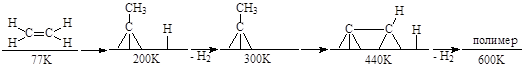
Реакции поверхностных интермедиатов включают стадии окислительного присоединения, восстановительного элиминирования, внедрения, a- и b-элиминирования, гидрогенолиза M-C и С-С связей и др. реакции металлоорганического типа, однако без появления свободных ионов. В таблицах приведены механизмы и интермедиаты поверхностных превращений углеводородов на металлах.
Таблица 3.1. Каталитические реакции, включающие разрыв С-С связи.
| Процесс |
Предполагаемый механизм |
| Гидрогенолиз |
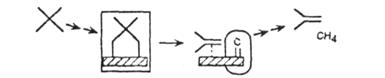 |
| Гидрогенолиз |
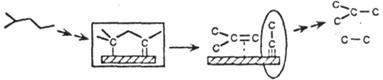 |
| Гидрокрекинг |
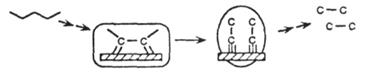 |
| Деметилирование |
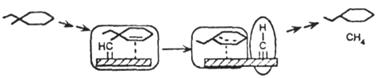 |
| Изомеризация алканов |
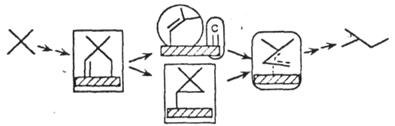 |
| Изомеризация/ дегидроциклизация алканов |
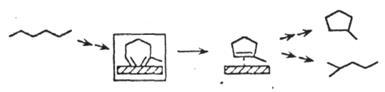 |
Обозначения:  – алкил, металлацикл; – алкил, металлацикл;
 – карбен, аллил; – карбен, аллил;
 – карбин, винил. – карбин, винил.
Таблица 3.2. Каталитические реакции, включающие образование С-С связи.
| Процесс |
Предполагаемый механизм |
| Дегидроциклизация |
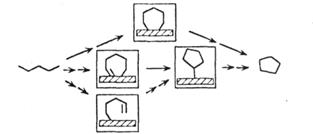 |
| Дегидроциклизация |
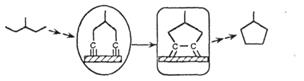 |
| Синтез Фишера-Тропша |
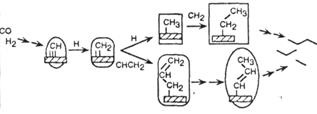 |
| Полимеризация олефинов |
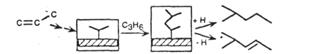 |
| Полимеризация ацетилена |
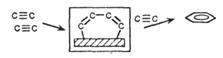 |
Обозначения: см. табл. 3.1.
Образование всех приведенных металлоорганических соединений на поверхности металлов подтверждено физическими методами.
Вопросы для самоконтроля
1) Как проявляется правило наименьшего изменения валентной оболочки металла в ходе ЭС в реакциях переноса электрона?
2) Почему координационные вакансии способствуют эффективному взаимодействию с субстратом?
3) Перечислить основные типы реакций координированных лигандов.
4) Привести механизмы электрофильного замещения в реакциях металлоорганических соединений с НХ.
5) Привести примеры поверхностных металлоорганических соединений.
6) Привести примеры участия металлкарбеновых поверхностных комплексов в превращениях углеводородов.
Литература для углубленного изучения
1. Темкин О.Н., Кинетика каталитических реакций в растворах комплексов металлов, М., МИТХТ, 1980, Ч.III.
2. Коллмен Дж., Хигедас Л., Нортон Дж., Финке Р., Металлоорганическая химия переходных металлов, М., Мир, 1989, т. I, т. II.
3. Моисеев И.И., p-Комплексы в окислении олефинов, М., Наука, 1970.
4. Темкин О.Н., Шестаков Г.К., Трегер Ю.А., Ацетилен: Химия. Механизмы реакций. Технология. М., Химия, 1991, 416 с., раздел 1.
5. Хенрици-Оливэ Г., Оливэ С., Координация и катализ, М., Мир, 1980, 421 с.
6. Крылов О.В., Матышак В.А., Промежуточные соединения в гетерогенном катализе, М., Наука, 1996.
7. Zaera F., An Organometallic Guide to the Chemistry of Hydrocarbon Moities on Transition Metal Surfaces., Chem. Rev., 1995, 95, 2651 – 2693.
8. Bent B.E., Mimicking Aspects of Heterogeneous Catalysis: Generating, Isolating, and Reacting Proposed Surface Intermediates on Single Crystals in Vacuum, Chem. Rev., 1996, 96, 1361 – 1390.
|
