Государственное образовательное учреждение
высшего профессионального образования
Московский Педагогический Государственный Университет
Химический факультет
Кафедра физической и аналитической химии
Курсовая работа
Тема:
«Моделирование адсорбции катионов тяжелых металлов на почве при различных значениях рН».
Чернова М.Н. студентка IV курса
Руководитель:
КХН, доцент
Казакова Г.Д.
КХН, доцент
Горичев И.Г.
Москва 2010год
Содержание:
- Введение
- Литературный обзор
- 1. Адсорбция на природных сорбентах
- 1.1 Основные теории адсорбции
- 1.2 Пористая структура и удельная поверхность адсорбента.
Активные центры и строение поверхностного слоя.
- 2. Место почвы в миграционной системе биосферы
- 2.1 Водонерастворимые формы
- 2.2 Гумусовые кислоты
- 3. Экспериментальная часть
- 3.1 Ход определения
- 3.2 Результаты и их обсуждения
- 3.3 Выводы
- Приложение
- Библиография
Введение
Почва, как многокомпонентная система состоит из таких фракции, как глинистая, иловая, песчаная, смешанная и другие. Практически все из них обладают высокой адсорбционной способностью,
Так глинистые минералы, благодаря высокой дисперсностью, а потому и чрезвычайно развитой поверхностью, обусловливают очень большую адсорбционную способность – активно поглощать из растворов различные вещества и химические элементы. Данное обстоятельство определяет важность и актуальность изучения взаимодействия глинистых минералов и других фракций почв с тяжелыми металлами.
Целью настоящей работы являлось исследование способности песка, глины, CuO, SiO2
, FeOOH к адсорбции ионов тяжелых металлов на примере железа, цинка, меди, и их десорбции, а также изучение зависимостей зарядов поверхности образцов песка, насыщенных этими металлами, от pH среды.
В связи, с чем решались следующие задачи:
1. потенциометрическое определение поверхностного заряда на песок, CuO, SiO2
, FeOOH.
2. исследование адсорбции ионов железа, цинка, меди на песке.
Литературный обзор
1. Адсорбция на природных сорбентах
Процессы адсорбции широко применяются в промышленности при очистке и осушке газов, очистке и осветлении растворов, разделении газов и паров. Адсорбцию применяют главным образом при невысоком содержании поглощаемого компонента в исходной смеси, когда требуется достичь практически полного извлечения адсорбтива. Этим объясняется возросшее в последнее время значение адсорбционных процессов для современного производства вследствие расширения потребности в веществах высокой чистоты.
Реклама

Кроме того, проблема переработки отходов материального производства и ассимиляции их в окружающей среде ставит задачу комплексного использования минерального сырья и создания безотходных технологий. Для разрешения этого вопроса требуется организация переработки и очистки промышленных выбросов. Их глубокую очистку способны обеспечить безреагентные методы, в первую очередь адсорбционные.
Перспективность использования адсорбентов основана на их высокой эффективности при извлечении компонентов промышленных выбросов, экологической чистоте, надежности, простоте в эксплуатации. Однако использование адсорбционных технологий обезвреживания крупнотоннажных промышленных стоков накладывают специфичные требования к используемым адсорбционно-активным материалам: они должны быть полифункциональными для обеспечения очистки стоков и выбросов в широком диапазоне их состава по технологии скорых фильтров. Не менее важным вопросом является последующая утилизация отработанного материала. Адсорбенты, синтезированные на основе кондиционного сырья, не отвечают этим требованиям. Они дефицитны, их применение требует создания определенных условий сорбции.
Эта проблема может быть решена использованием более дешевых адсорбционно-активных материалов, полученных на основе природных минеральных сорбентов.
Природные минералы, обладающие высокой адсорбционной активностью и используемые для получения промышленных сорбентов, условно можно разделить на два класса: горючие ископаемые (каменный и бурый уголь, сланцы, торф) и природные алюмосиликаты (цеолиты, бентониты, каолины).
К алюмосиликатам, имеющим существенную адсорбционную способность, относят глинистые минералы и цеолиты.
Цеолиты как осадочного, так и вулканического происхождения, благодаря особенностям кристаллического строения, имеют более предпочтительные для условий материального производства адсорбционные свойства, чем глины. Основные направления использования природных цеолитов – адсорбенты воды и органических соединений, катализаторы, ионообменные материалы для поглощения ионов тяжелых металлов.
Природные глинистые минералы уступают по адсорбционным характеристикам цеолитам. Однако наличие крупнейших залежей осадочных пород в центральной части и на юге России, дешевизна разработки обуславливают возможность применения их в качестве адсорбционно-активных материалов и получения на их основе промышленных адсорбентов.
Реклама

1.1 Основные теории адсорбции
Адсорбция – это концентрирование компонента в поверхностном слое, по сравнению с объемной фазой.
Явление адсорбции обусловлено избытком поверхностной энергии, возникающим вследствие нескомпенсированного действия межмолекулярных сил в поверхностном слое на границе фаз. Величину избытка энергии, называемой поверхностной энергией Гиббса, определяют как произведение поверхностного натяжения s (фактор интенсивности) и площади поверхности раздела фаз s(фактор емкости).
В многокомпонентных системах в адсорбированное состояние предпочтительнее перейдет тот компонент смеси, который сильнее уменьшит межфазное натяжение.
В процессе адсорбции при перераспределении компонентов между поверхностным слоем и объемной фазой происходит изменение их химических потенциалов, поэтому процесс можно также рассматривать как переход поверхностной энергии в химическую. Соотношение между поверхностным натяжением и химическим потенциалом компонентов системы показано фундаментальным адсорбционным уравнением Гиббса:
-ds = SGi
dmi
,
где Gi
=hi
/s– поверхностный избыток адсорбата компонента; в поверхностном слое по сравнению с его равновесной концентрацией в объемной фазе так называемая гиббсовская адсорбция:
G=V (c0
i
-ci
)/s,
где V – объем фазы, м3
;
ci
– равновесная объемная концентрация i-го компонента;
c0
i
- исходная объемная концентрация, моль/ м3
;
mi
– химический потенциал i-го компонента, моль/ м2
.
Согласно правилу фаз Гиббса число степеней свободы для трехкомпонентной системы (адсорбент, адсорбтив, объемная фаза) равно трем, поэтому для количественной оценки равновесного состояния системы необходимо учитывать следующие параметры:
t – температура процесса; p(c) – парциальное давление пара или газа в объемной фазе или его концентрация; a – адсорбционная емкость адсорбента, количество адсорбата, поглощенного адсорбентом при конкретных условиях.
Для удобства изображения количественных соотношений в состоянии равновесия, один из трех параметров считают постоянным. Поэтому уравнения адсорбционного равновесия записываются тремя закономерностями:
изотермы адсорбции t= const:
1) изотермы адсорбции t= const:
a= ft
(c)= ft
`
(p);
2) изобары (изопикны) адсорбции p= const, c= const
a= fc
(T)= fp
`
(T);
3) изостеры адсорбции a= const
c= fa
(T)= fa
`
(T)
Построение единой теории адсорбции – задача чрезвычайно сложная, нерешенная до настоящего времени. Разработано большое количество теорий, позволяющих рассчитать с определенной долей вероятности величину адсорбции при заданных температуре и парциальном давлении адсорбата [7,11].
1.2 Пористая структура и удельная поверхность адсорбента.
Активные центры и строение поверхностного слоя.
Фактором емкости избыточной поверхностной энергии Гиббса является поверхность раздела фаз. Следовательно, для обеспечения эффективности процесса адсорбции эта поверхность должна доставляться пористостью твердых тел.
Поры твердого тела имеют решающие значения во многих технологических процессах: при фильтрационном разделении различных веществ, при регулировании физических свойств различных полимеров, для обеспечения прочностных свойств стройматериалов, при добыче каменных углей.
В соответствии с областью исследований разработано несколько систем классификации пор.
Фундаментальные теоретические исследования по влиянию пористой структуры адсорбентов на их адсорбционную способность проведены академиком Дубининым М.М. и его учениками. Ими установлена явная зависимость адсорбционных свойств различных адсорбентов от характера пористости, которая позволила разработать научно обоснованную классификацию пор и адсорбентов по их структурным типам, принятую Международным союзом чистой и прикладной химии (ИЮПАК).
Классификация отражает характер адсорбционных процессов, протекающих в порах. Основные параметры этой классификации представлены в таблице 1.
Таблица 1.
Классификация пор (по Дубинину М.М.)
| Название пор |
размеры пор |
роль в процессе адсорбции |
| радиус, Ао
|
удельная поверхность, Sуд.
, м2
/г |
суммарный объем пор V∑
, см3
/г |
| макропоры |
>1000-2000 |
0,5-2,0 |
0,2-0,8 |
играют незначительную роль в статике адсорбции, оказывают существенное влияние на кинетику, играют роль транспортных каналов |
| переходные поры |
15 (1000-2000) |
10-400 |
- |
в переходных порах происходит послойная адсорбция, завершается капиллярной концентрацией |
| микропоры |
<15 |
500-1000 |
<0,5*
|
в микропорах адсорбция носит характер объемного заполнения |
*для промышленных микропористых сорбентов
В соответствии с этой классификацией и в зависимости от преимущественного процесса адсорбции Дубинин М.М. определил три типа пористых тел: микропористые, макропористые и переходнопористые.
На эффективность адсорбции влияет не только геометрическая характеристика, но и их структура, т.е. комбинация микро-, макро- и переходных пор.
Твердое тело, обладающее тонкими порами, не может быть эффективным сорбентом, если не имеет развитой структуры транспортных переходных и макропор.
Кроме структуры и размеров большое значение имеет форма пор.
Центры физической адсорбции распространяются во впадинах, трещинах, зазорах, где действие межмолекулярных сил возрастает с увеличением молекулярного окружения. Выступы, ребра, углы несут атомы с меньшим числом соседей. Они обладают большим числом ненасыщенных химических связей и выполняют роль центров хемосорбции.
В качестве поверхностного центра рассматривается микроскопическая группа атомов (или один атом), которая в каком-либо смысле химически активна.
Решетки выделяют кислотные центры Льюиса L+
, способные присоединять e-
и кислотные центры Бренстеда, обладающие тенденцией отдавать протон (рис.1). При этом один вид центров может переходить в другой, а прочность связей, хемосорбированных на поверхности молекул воды, определяет кислотные или основные свойства сорбента.
-
H+
H – O
a) – Si – O – Al – O – Si – б) – Si – O – Al – O – Si –
OO
– Si – – Si –
Рис. 1 Кислотные активные центры на поверхности алюмосиликатов:
а) – кислота Льюиса (атом алюминия, имеющий три связи с кислородом кремнекислородного состава, требует дополнительную электронную пару для заполнения p-орбитали);
б) – кислота Бренстеда (образована из льюисовского центра, который координационно связал молекулу воды, ставшую источником протонов).
Активными центрами ионного обмена являются подвижные катионы.
Химический состав поверхностного слоя является природой твердого материала.
Так как объектом исследований являются алюмосиликаты, то достаточно рассмотреть строение поверхности кремнезема.
Поверхность кремнезема устилают силанольные –Si–OH и силоксановые –Si–O–Si– группы, соотношение которых может изменяться в зависимости от степени дегидроксилирования.
Образование поверхностного гидроксильного слоя на сколах кристаллической решетки обусловлено насыщением разорванных связей кристаллов в результате диссоциации воды: к каждому атому кислорода присоединяется водород, а к атомам металла – гидроксильные группы. Кислород OH-группы стремится образовать связь с несколькими атомами металла, если их взаимное расположение не препятствует этому. При таком построении атомы кислорода поверхностных OH-групп всегда располагаются в тех местах, где должны находиться атомы кислорода в бесконечной кристаллической решетке.
Известно, что при повышении температуры прогрева кремнезема силанольные группы способны конденсироваться, образуя силоксановые группы, при этом сначала удаляется физически адсорбированная вода (t<120o
C), а затем происходит конденсация силанольных групп (180-400o
C). Однако эти процессы могут происходить одновременно.
Силанольные группы значительно более активны и легче вступают в химические реакции, чем силоксановые, так как протон силанольной группы имеет слабокислый характер и способен вступать в реакции обмена.
По данным Снайдера на поверхности кремнезема в различных соотношениях может находиться пять видов групп:
H
OHOH…O – HO
а) – Si – б) – Si – в) – Si – O – Si –
OHO – H…O
г) – Si – OH д) – Si – O – Si –
а) силанольная (связанная вода – свободные, отдельно стоящие OH-группы);
б) физически связанная вода – молекулы воды, имеющие водородные связи с силанольными группами;
в) дегидратированные оксиды – силоксановые группы;
г) близнецовые (геминальные) группы OH, связанные с одним атомом кремния;
д) реакционноспособные вицинальные группы OH, преобладающие в тонкопористых кремнеземах – соседние близкорасположенные OH-группы, связанные между собой водородной связью.
Такое строение поверхности кремнезема объясняет значительный вклад водородной связи в адсорбционное взаимодействие между адсорбатом и адсорбентом силикатного и алюмосиликатного типа [7].
2. Место почвы в миграционной системе биосферы
Среди миграционных процессов в биосфере наиболее изучен так называемый биологический круговорот, под которым подразумевается циклический обмен масс химических элементов
почвы и растительности. Количество металлов, участвующих в биогеохимической миграции,
внушительно: массы цинка и меди, ежегодно вовлекаемые в такой круговорот на всей площади
мировой суши, оцениваются миллионами тонн, никеля и свинца — сотнями тысяч, ртути и кадмия — тысячами тонн. Другой мощный массопоток тяжелых металлов обусловлен водным стоком. Атмосферные осадки, промывая почвенную толщу и смывая с ее поверхности мелкие частицы, одновременно вовлекают в водную миграцию тяжелые металлы. Количество некоторых из них, выносимое реками на протяжении года, столь велико, что превышает их мировую добычу. Но не вся масса металлов может быть за хвачена растительностью в биологический круговорот. Если бы их остаточные количества не удалялись за пределы суши, то в течение непродолжительного времени наземные экосистемы переполнились активными формами тяжелых металлов, что имело бы пагубные последствия для живых организмов. В миграционной системе биосферы особое место занимает почва, в которой зарождаются главные массопотоки металлов. С одной стороны, здесь происходит мобилизация тяжелых металлов, находящихся в рассеянном состоянии, с другой перераспределение их масс, непрерывное высвобождение из растений, микроорганизмов, разрушающихся горных пород. Благодаря равновесию между физико-химическими условиями и различными формами нахождения рассеянных металлов, а также их способности включаться в ту или иную миграцию, не только поддерживаются миграционные потоки, но и регулируется их накопление. Избыточное количество металлов путем трансформации их форм выводится в твердую фазу почвы, где они могут концентрироваться и в дальнейшем пополнять отдельные миграционные потоки. Для понимания механизмов мобилизации и саморегуляции масс металлов при водном перемещении, прежде всего, необходимо изучить их миграционные формы, большая часть которых образуется в почве.
2.1 Водонерастворимые формы
Как ни важна миграция водорастворимых форм, основная масса тяжелых металлов, выносимых речным стоком, связана с водонерастворимыми соединениями. Минеральная часть почвысостоит из компонентов двух фракций: мелкообломочной и высокодисперсной. Граница междуними проходит в интервале 0,002—0,004 мм. Здесь могут присутствовать как наиболее мелкиеобломочные компоненты, так и наиболее крупные из высокодисперсных. Среди мелкообломочных, как правило, преобладает кварц — наиболее устойчивый к воздействию процессов почвообразования и выветривания. Другие обломочные минералы — полевые шпаты, слюды, железомагнезиальные силикаты, кремни — обычно не превышают 10%. Количество тяжелых металлов, содержащихся в этой фракции крайне незначительно, меньше, чем впочве в целом. Они входят в кристаллическую структуру минералов и освобождаются по мере ихразрушения. Высокодисперсная фракция состоит из минералов, обладающих слоистой кристаллическойструктурой, в которой слои соединены между собой менее прочно, чем ионы в структурах обломочных минералов. Такая особенность данных минералов способствует сорбции рассеянных тяжелых металлов, и концентрация их в этой фракции выше, чем в мелкообломочной и в почве вцелом. Высокодисперсная фракция обычно изучается методом рентгеноструктурного анализа. В еесостав входят минералы группы смектитов, сильно набухающие при насыщении их этиленгликолем и сжимающиеся при нагревании до 550°С; гидрослюды, межплоскостные расстояния у которых остаются стабильными при насыщении этиленгликолем и при нагревании, и минералы группы каолинита с пониженной способностью к сорбции. Члены второй и особенно первой группактивно фиксируют ионы металлов и их органические соединения. Детальное изучение фракционного состава высокодисперсных частиц показало, что в нижних горизонтах почвенного профиля преобладают частицы размером от 0,8 до 1,5 мкм. При почвообразовании происходит их измельчение и аморфизация. Поэтому в верхнем (гумусовом) горизонте почв сильно возрастает содержание рентгеноаморфных частиц размером менее 0,1 мкм. С помощью растрового электронного микроскопа, при увеличении более чем в 3000 тыс.раз, можно видеть, что чешуйки глинистых минералов размером 0,8— 1,2 мкм образуют субпараллельные срастания, которые агрегированы в сильно пористую массу. Матовая поверхностьотдельных чешуек и их срастаний говорит о том, что на них накапливаются аморфные вещества,по-видимому, водонерастворимые гумусовые соединения.В черноземно-степных почвах срастания сильно агрегированы, в микропустотах присутствует субкристаллический кальцит. В почвах подзолистого типа глинистые минералы агрегированы в меньшей степени, а в порах нередко наблюдаются скопления рентгеноаморфных гидроксидов трехвалентного железа.
2.2 Гумусовые кислоты
Важная роль в структурной организации минерального вещества принадлежит активной
части почвенного гумуса — гумусовым кислотам. При этом действие двух главных групп гумусовых кислот — фульвовых и гуминовых, — обладающих неодинаковой растворимостью, существенно различается. Ионы металлов, сорбированные на поверхности почвенных частиц, могут образовыватькомплексные соединения с фульвокислотами и в этой форме переходить в раствор. Водонерастворимые гуминовые кислоты еще более активно соединяются с тяжелыми металлами и выводят их из раствора в твердую фазу почвы. В свою очередь, гуминовые кислоты
сорбируются высокодисперсными минеральными частицами, и тяжелые металлы оказываются закрепленными в их пленках и сгустках. Подобные пленки склеивают отдельные частички с образованием микроагрегатов. В прозрачных шлифах, приготовленных из почвы без нарушения ее строения, хорошо видно, что желто-бурые пленки и сгустки пропитывают минеральное вещество. Их часто принимают за гидроксиды железа. Но с помощью иммерсионного метода под микроскопом легко устанавливается, что показатель преломления бурых пленок значительно ниже, чем гидроксидов железа, и соответствует показателю преломления гуминовых кислот. Вместе с тем, благодаря высокому содержанию железа в почве, его концентрация в гуминовых пленках не сравнимо больше, чем других металлов. Гуминовые кислоты с разной молекулярной массой и растворимостью отличаются взаимодействием с почвенными минералами [8, 9]. Их структурные соотношения с высокодисперсными минеральными частицами сложны и недостаточно изучены. Одна из моделей связи молекулы гуминовой кислоты с обладающим кристаллической структурой дисперсным минералом и коллоидной частицей показана на рисунке.
Миграционные формы металлов, образованные разными группами (фракциями) гуминовых кислот, также различаются по свойствам, что отражается на формировании миграционных потоков. В частности, почвенные частицы, сорбированные низкомолекулярными гуминовыми кислотами, быстрее дезагрегируются и легче переходят в состав высокодисперсных речных взвесей. Чтобы понять закономерности водной миграции тяжелых металлов в биосфере, необходимо провести сравнительное изучение водонерастворимых миграционных форм тяжелых металлов в почвах главных природных зон мира.
3. Экспериментальная часть
Определение заряда поверхности методом потенциометрического титрования
В литературе описано применение метода потенциометрического титрования для изучения кислотно-основных свойств оксидов. При контакте оксидов металлов с раствором электролита происходит адсорбция или десорбция потенциалопределяющих ионов Н+
, образующихся в том числе и в результате диссоциативной хемосорбции воды.
Адсорбция потенциалопределяющих ионов всегда сопровождается адсорбцией катионов и анионов электролита. В результате устанавливаются кислотно-основные равновесия, которые определяют заряд поверхности оксида и скачок потенциала в ионной части двойного электрического слоя.
Согласно модели кислотно-основных равновесий на границе оксид/раствор, можно выделить четыре адсорбционных равновесия:
MOH+
2,s
<-> MOH0
s
+ H+
MOH0
s
<-> MO-
s
+ H+
MOH0
s
+ H+
+Cl-
<-> MOH+
2,s
…Cl-
MOH0
s
+K+
<-> MO-
s
…K+
+ H+
,
где индексом s указаны частицы на поверхности оксида, принимающие участие в адсорбционном равновесии. Заряд поверхности (q) возникает за счет адсорбированных на поверхности оксида [MOH+
2,
s
] и [MO-
s
], [MOH+
2,
s
…Cl-
] и [MO-
s
…K+
] групп: q = ([MOH+
2,
s
…Cl-
] + [MOH+
2,
s
]) – ([MO-
s
…K+
] + [MO-
s
]).
[MOH+
2,
s
], [MO-
s
], [MOH+
2,
s
…Cl-
], [MO-
s
…K+
] - поверхностные концентрации частиц, выраженные в Кл/см2
, [Н+
], [К+
], [Сl-
] - концентрации ионов в объеме раствора.
3.1 Ход определения.
Для химического анализа готовят навески: чистого песка; песка, насыщенного ионами железа, цинка и меди; CuO, SiO2
, FeOOH.
Измерение pH проводили на pH-метре, который проверялся по буферным растворам. В ходе эксперимента в стакан на 100 мл наливают по 25 мл растворов 0,1 м HCl и 0,01 н KCl, добавляют навеску песка, CuO, SiO2
, FeOOH, массой 5 г. При перемешивании с помощью магнитной мешалки титруют 1 м раствором KOH. Измерение pH записывают после каждого добавления щелочи по 0,1 мл до значения pH=12.
Используя полученные значения pH, с помощью компьютерной программы находят значения зарядов поверхности, после чего строят кривые зависимости зарядов поверхности от pH.
3.2 Результаты и их обсуждения
Результаты адсорбционных исследований представлены в табл. 2, 3, 4 и на соответствующих им рис. 2, 3, 4, из которых видно как менялись концентрации металлов в ходе экспериментов для трех различных исходных концентраций – 1, 5 и 10 мг/л.
Таблица 2
Результаты взаимодействия песка с железом
| Исходная концентрация железа, мг/л |
Количество железа, оставшегося в растворе, мг/л |
Количество железа, адсорбированного песком, мг/л |
Количество железа, перешедшего в раствор после десорбции, мг/л |
Количество железа, оставшегося на песке после десорбции, мг/л |
| 1 |
0,01 |
0,99 |
0,2 |
0,79 |
| 5 |
0,03 |
4,97 |
0,17 |
4,8 |
| 10 |
0,8 |
9,2 |
0,15 |
9,05 |
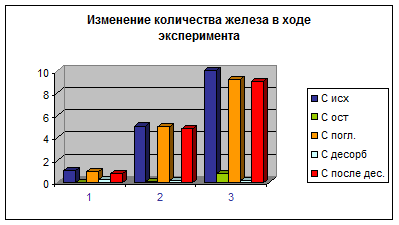
Рис. 2 Изменение количества железа в ходе эксперимента
Таблица 3
Результаты взаимодействия песка с цинком
| Исходная концентрация цинка, мг/л |
Количество цинка, оставшегося в растворе, мг/л |
Количество цинка, адсорбированного песком, мг/л |
Количество цинка, перешедшего в раствор после десорбции, мг/л |
Количество цинка, оставшегося на песке после десорбции, мг/л |
| 1 |
0,5 |
0,5 |
0,5 |
0 |
| 5 |
4,1 |
0,9 |
0,83 |
0,07 |
| 10 |
9 |
1 |
1 |
0 |
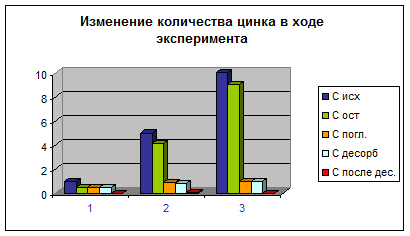
Рис. 3 Изменение количества цинка в ходе эксперимента
Таблица 4
Результаты взаимодействия песка с медью
| Исходная концентрация меди, мг/л |
Количество меди, оставшейся в растворе, мг/л |
Количество меди, адсорбированной песком, мг/л |
Количество меди, перешедшей в раствор после десорбции, мг/л |
Количество меди, оставшейся на песке после десорбции, мг/л |
| 1 |
0,9 |
0,1 |
0 |
0,1 |
| 5 |
0,81 |
3,19 |
0,81 |
2,38 |
| 10 |
0,35 |
8,65 |
0,72 |
7,93 |

Рис. 4 Изменение количества меди в ходе эксперимента
На рис. 5 рассматриваются изменения остаточной концентрации ионов тяжелых металлов на песке для трех различных исходных концентраций – 1, 5 и 10 мг/л. Остаточная концентрация характеризует количество металла, не поглощенного песком после проведения эксперимента по адсорбции.

Рис. 5 Сравнение остаточных концентраций металлов
Очень важно при обсуждении результатов учесть процесс десорбции, т.к. от него зависит эффективность или не эффективность проделанной работы. Сравнение концентрации десорбированного металла изображено на рис. 6. Из него видно, что минимальное сродство к десорбции у железа и наибольшее у цинка.

Рис. 6 Способность металлов к десорбции
Также проверяются остаточные концентрации ионов металлов на песке после десорбции, что отображено на рис. 7. Из чего следует, что легче всего десорбируется цинк, т.к. сам процесс адсорбции для него менее характерен. Наименьшие значения по десорбции у железа - настолько велико его сродство к адсорбции.

Рис. 7 Остаточное количество металла на песке после десорбции
По результатам эксперимента четко видна зависимость способности к адсорбции от типа металла. Из рис. 8 следует, что железо адсорбируется лучше меди, а медь в свою очередь обладает большим адсорбционным сродством, чем цинк. Полученные данные хорошо согласуются со многими литературными источниками.
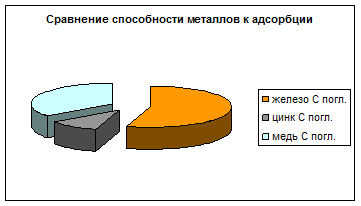
Рис. 8 Сравнение способности металлов к адсорбции
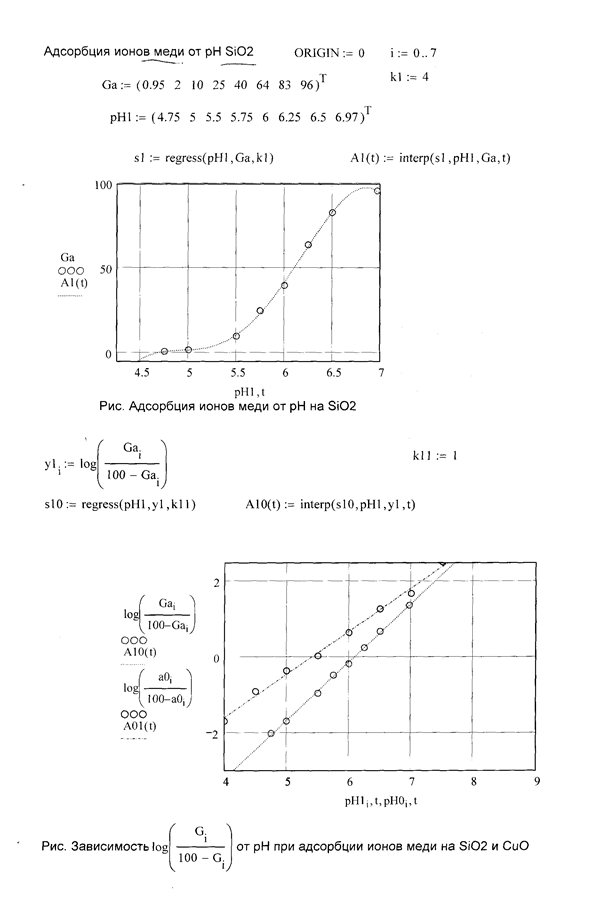
Также были изучены кислотно-основные характеристики природного образца диоксида кремния.
Проведены исследования по изучению зарядов поверхности различных образцов песка с использованием метода потенциометрического титрования. Песок был обработан растворами, содержащими ионы тяжелых металлов. В качестве рассматриваемых металлов были выбраны: железо(III), цинк(II) и медь(II).
Результаты представлены на рис. 9, на котором видны точки нулевых зарядов, т.е. такие значения pH, при которых заряд поверхности равен нулю (концентрации ионов водорода и гидроксильных ионов одинаковы). По ним можно судить о кислотных или основных свойствах образца.
Для природного образца диоксида кремния, ничем не обработанного, такое значение pH равно четырем. Более кислая природа у песка, обработанного раствором, содержащим ионы железа (pH0 = 1,95). У образцов, обработанных растворами, содержащими ионы цинка и меди, слабо-щелочные и нейтральные свойства (pH0 = 7,48 и pH0 = 6,78 соответственно). Эти данные подтверждают ряд адсорбции ионов тяжелых металлов, полученный в адсорбционных исследованиях.

Рис. 9 Зависимость зарядов поверхности от pH для чистого песка и песка, насыщенного ионами железа, цинка, меди
3.3 Выводы
1. Установлено, что адсорбция ионов тяжелых металлов зависит от концентрации ионов и pH среды и убывает в следующем ряду:
Fe3+
> Cu2+
> Zn2+
.
2. Показано, что центры адсорбции диоксида кремния проявляют кислотно-основные свойства. Поверхность исследуемого диоксида кремния имеет кислотную природу. pH нулевого заряда (pH0) = 4.
3. Величина нулевого заряда диоксида кремния, обработанного растворами, содержащих ионы железа, цинка, меди изменяется следующим образом:
pH0 (Zn2+
) > pH0 (Cu2+
) > pH0 (Fe3+
).
Приложение


Библиография:
1. Березюк В.Г. Изучение сорбционных свойств природных алюмосиликатов. – Екатеринбург.
2. Варшал Г.М. Формы миграции фульвокислоты и металлов в природных водах. – Москва: изд.-во МИР, 1994, -40с.
3. Варшал Г.М., Кощеева И.Я., Сироткина И.С. // Геохимия. 1979. №4. С.598—607.
4. Веницианов Е.В., Лепихин А.П. Физико-химические основы моделирования миграции и трансформации тяжелых металлов в природных водах. – Екатеринбург: изд.-во РосНИИВХ, 2002, -236с.
5. Дривер Дж. Геохимия природных вод. Москва: изд.-во МИР, 1985,-440с.
6. Линник П. Н., Набиванец Б. И. Формы миграции металлов в пресных поверхностных водах. – Ленинград, 1986, -272с.
7. Минералы. Силикаты со структурой, переходной от цепочечной к слоистой. Справочник. Том IV. Выпуск 1. – Москва, 1992
8. Орлов Д.С. Гумусовые кислоты почв и общая теория гумификации. М., 1990.
9. Тарасевич Ю.И., Овчаренко Ф.Д. Адсорбция на глинистых минералах. – Киев, 1975
10. Тейт Р. Органическое вещество почвы. М., 1991.
11. Фролов Ю.Г. Курс коллоидной химии. – Москва: изд.-во Химия, 1989, - 464с.
12. Шварценбах Г., Флашка Г. Комплексонометрическое титрование. – Москва: изд.-во Химия, 1970, -360с.
13. Электрохимия: определение констант кислотно-основного равновесия на границе оксид/раствор методом потенциометрического титрования. – Москва, 1993, том 29, №3, -310-313с.
|
