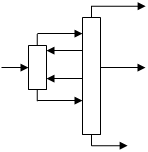содержание
1.Введение.
2.Литературный обзор.
2.1.Методы разделения азеотропных смесей.
2.1.1. Разделение в колоннах работающих под разным давлением
2.1.2. Азеотропная ректификация
2.1.3. Теоретические основы экстрактивной ректификации
2.2.Экстракционное выделение аренов из катализатов риформинга бензиновых фракций.
2.2.1.Экстракция аренов С6
-С8
сульфоланом из катализатов риформинга.
2.3. Процесс обратимой ректификации, как процесс обладающий минимальной энтропией.
2.4. Способы приближения необратимых процессов ректификации к процессу обратимой ректификации.
2.4.1. Колонны со стриппинг-секциями.
2.4.2. Многокорпусная ректификация.
2.3.3. Теплоинтеграция потоков.
2.4.4. Совершенствование массообменных устройств.
2.5. Методы воздействия на эффективность работы ректификационных колонн.
2.6. Ректификация многокомпонентных смесей.
2.7. Поливариантность технологических схем.
2.8. Термодинамические методы расчета равновесия жидкость-пар.
2.8.1. Модели локальных составов [12].
2.8.2 Уравнения состояния[12].
2.8.3 Групповые модели[12].
3. Постановка задачи
4. Расчетно–экспериментальные методики и аппаратные средства
4.1 Программный комплекс PRO/II.
4.1.1. Методы расчета ректификации.
4.2. Синтез технологических схем экстрактивной ректификации в комплексах простых и сложных колонн.
5. Расчетно-экспериментальная часть
5.1. Физико – химические свойства индивидуальных компонентов.
5.2. Моделирование паро-жидкостного равновесия
Список литературы.
Процессы разделения многокомпонентных смесей органических продуктов являются одними из самых распространенных и сложных процессов химической и нефтехимической технологии. Они используются как на стадиях предварительной подготовки сырья, так и непосредственно в общей технологической схеме производства для разделения полупродуктов и получения продуктов высокой степени очистки. Эти процессы являются одними из самых энергоемких, и их эффективность часто определяет экономику производства в целом.
Ректификация наиболее часто применяемый метод для разделения продуктов и подготовки сырья в химической технологии. Она проста и дает возможность получить очень чистый продукт, однако при всех ее достоинствах, есть и ряд серьезных недостатков. Ректификационные колонны – это большие и металлоемкие аппараты, требующие крупных капиталовложений, более того это еще и энергоемкий процесс. На ректификацию может затрачиваться до 70% всей энергии производства. Поэтому становится насущной проблема разработки наиболее экономичной технологии разделения продуктов. При увеличении количества разделяемых компонентов увеличивается и число вариантов их разделения, отличающиеся друг от друга энергозатратами.
Реклама

Такие особенности производственных процессов как непрерывность и многотоннажность приводят к тому, что даже относительно невысокие снижение энергозатрат, повышение качества товарных фракций обеспечивают значительный экономический эффект для технологии в целом. Существующие эвристические и алгоритмические методы поиска структуры оптимальной технологической схемы ректификации, как правило, ориентированы на выбор наилучшего решения в определенном заданном классе эквивалентности технологических схем.
В случае разделения азеотропных смесей получение чистых продуктов обычной ректификацией невозможно. Для решения этой проблемы предложен ряд методов, одним из которых является экстрактивная ректификация с использованием разделяющего агента. Данный метод достаточно прост в аппаратурном оформлении и не требует значительных энергозатрат по сравнению с другими методами разделения азеотропных смесей.
2.1.Методы разделения азеотропных смесей.
В различных отраслях промышленности используют разнообразные жидкие и газовые смеси, подлежащие разделению на чистые компоненты или фракции различного состава. Разделение таких смесей проводят в комплексах, основанных на процессеректификации.
Необходимо иметь в виду, что смеси, образующие азеотроп, разделить на практически чистые компоненты методом обычной ректификации нельзя. В данном случае необходимо использовать специальные методы ректификации, такие как:
· разделение в колоннах, работающих под разным давлением;
· азеотропную ректификацию;
· экстрактивную ректификацию.
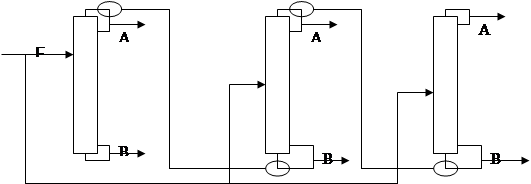
Для разделения азеотропных смесей путем обычной ректификации используются комплексы (рис.1),
работающие под разным давлением, которые позволяют преодолеть ограничения физико-химического характера и получать продукты требуемой чистоты. При этом используется свойство изменения состава азеотропной смеси с изменением температуры [1]. Составы азеотропов при разных давлениях различны, причем в зависимости от давления состав питания может принадлежать то одной, то другой области ректификации. Именно это свойство используется в двухколонных комплексах, предназначенных для разделения азеотропных смесей, в которых колонны работают при разных давлениях.
Реклама

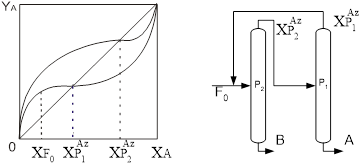
Рис.1. Разделение азеотропных смесей в комплексах, работающих под разным давлением
Путем изменения внешнего давления можно передвинуть азеотропный состав в область концентраций, отвечающих практически приемлемой чистоте одного из компонентов системы, и тогда ректификация на практически чистые компоненты окажется уже возможной. Изменение состава азеотропа, приходящееся на один градус температуры, согласно правилу Вревского, зависит от разности молярных теплот испарения компонентов. Следовательно, рассматриваемый метод разделения тем эффективнее, чем больше отличаются теплоты испарения компонентов разделяемой смеси.
Обычно теплоты испарения значительно различаются у веществ различной химической природы. Но практическое применение метода ограничено, т.к. возможный интервал изменения давлений ограничен температурами хладагентов, используемыми для конденсации паров в дефлегматорах, и теплоносителей, применяемых в кубах ректификационных установок. В силу указанных причин метод ректификации при двух различных давлениях не получил широкого практического применения для разделения азеотропных смесей.
Достаточно высокая степень разделения жидких смесей на компоненты может быть достигнута путём ректификации. Разделение обычно осуществляется в колонных аппаратах при непрерывном контакте фаз. Проводя последовательно ряд процессов испарения жидкости и конденсации пара, можно получить в итоге жидкость (дистиллят), представляющую собой практически чистый низкокипящий компонент (НКК). Аналогично, исходя из паровой фазы с соответствующим составом жидкости, путём проведения ряда последовательных процессов конденсации и испарения можно получить жидкость (кубовый остаток), состоящую почти из чистого высококипящего компонента (ВКК).
Процесс ректификации осуществляется путём многократного контакта между неравновесными жидкой и паровой фазами, движущимися относительно друг друга. При взаимодействии фаз между ними происходит массо- и теплообмен, обусловленные стремлением системы к состоянию равновесия. В результате каждого контакта компоненты перераспределяются между фазами. Многократное контактирование приводит к практически полному разделению исходной смеси.
Таким образом, отсутствие равновесия при движении фаз с определённой скоростью и многократность их контактирования являются непременным условием проведения ректификации.
Метод азеотропной ректификации (АР) основывается на проведении процесса ректификации с разделяющими агентами, обладающими свойством либо разбивать азеотроп, либо образовывать с одним или несколькими компонентами исходной смеси азеотропные системы и тем самым увеличивать коэффициенты относительной летучести разделяемых компонентов [2]. Процессы азеотропной ректификации стараются проводить так, чтобы вводимый в колонну разделяющий агент полностью выводился с дистиллатом. В виде кубового продукта можно получить один компонент или смесь нескольких компонентов с минимальным содержанием разделяющего агента (РА).
Так как в этом процессе РА выводится из системы в виде азеотропных смесей, его регенерация представляет большие трудности. В связи с этим наиболее желательными являются РА, обладающие ограниченной взаимной растворимостью в компонентах, отбираемых в виде дистиллата. В этом случае рецикл РА может быть осуществлен путем расслаивания охлажденного конденсата, отбираемого из верха колонны, и отбора в качестве дистиллата слоя, обогащенного целевым веществом. Содержащийся в этом растворе РА может быть отогнан в регенерационной колонне в виде азеотропа, также подвергаемого расслаиванию после конденсации и охлаждения.
Практический чистый целевой компонент получается в регенерационной колонне в виде кубовой жидкости. Типичная схема процесса азеотропной ректификации в присутствие РА представлена на рис.2
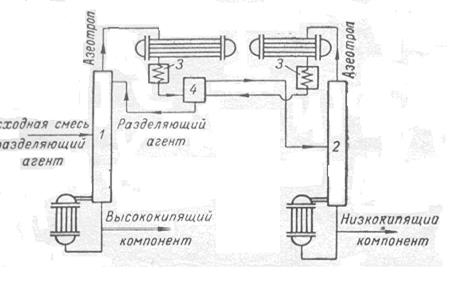
Рис. 2 Схема установки для азеотропной ректификации
1 –ректификационная колонна; 2- колонна регенерации РА; 3 – холодильник; 4 – расслаиватель.
2.1.3. Теоретические основы экстрактивной ректификации
Экстрактивная ректификация (ЭР) весьма часто применима в промышленности и она становится все более и более значимым методом разделения в нефтехимической инженерии. Масштаб отбираемого продукта на промышленном оборудовании варьируется от нескольких килотонн (диаметр колонны около 0,5 м) до сотен килотонн (диаметр колонны около 2,5 м). Процесс главным образом используется в следующих случаях: одним из применений является разделение углеводородов с близкими значениями температур кипения, таких как смеси состава С4
, С5
, С6
и т.д. Другое – это разделение смесей, образующих азеотроп, например спирт-вода, ацетокислота-вода, ацетон-метанол, метанол-метил-ацетат и т.д.
В ЭР добавляемый растворитель, т.е. разделяющий агент (РА), используется для изменения относительной летучести разделяемых компонентов. Таким образом возможно получить в одной колонне один чистый компонент с верха, а второй вместе с растворителем с низа, которые могут быть легко разделены во второй дистилляционной колонне, благодаря высокой температуре кипения растворителя. В процессе экстрактивной ректификации не обязательно выпаривать растворитель. Тогда как в азеотропной ректификации и растворитель и компоненты должны быть выпарены на верх азеотропной колонны ректификации. Более того, количество растворителя, применяемого в процессе азеотропной ректификации, обычно весьма велико. Это приводит к большему расходу энергии по сравнению с экстрактивной ректификацией. По этой причине, последняя является более предпочтительней. Недавно был предложен особый метод экстракции, адсорбционная экстракция, который представляет некий интерес. [3]
В общем, экстрактивная ректификация является одним из случаев реализации принципа перераспределения полей концентраций. При этом с одной стороны, преобразуется концентрационное пространство за счет добавления одного или нескольких экстрактивных агентов, которое обладает новым фазовым портретом по сравнению с исходным. С другой стороны, за счет разновысотной подачи экстрактивного агента и исходной смеси преобразуется динамическая система ректификации. Последнее порождает экстремумы на температурном профиле, соответствующем распределению компонентов по высоте колонны, что свидетельствует, о наличии элементов обратной ректификации[i]
,. Разновысотная подача потоков в колонну является обязательным условием реализации принципа перераспределения в данном методе.
В процессах экстрактивной ректификации регенерация разделяющего агента чаще всего не представляет затруднений. В связи с большим различием относительной летучести компонентов заданной смеси и разделяющего агента его регенерация легко осуществляется путем обычной ректификации, в процессе которой он отбирается в виде кубовой жидкости и вновь подается в колонну для экстрактивной ректификации. Традиционный комплекс экстрактивной ректификации состоит из двух ректификационных колонн: экстрактивной и колонны регенерации экстрактивного агента (ЭА). Такой комплекс представлен на рис.(3), где первая колонна является экстрактивной, куда подается тяжело- кипящий разделяющий агент, а сверху отбирается один из азеотропообразующих компонентов; продуктами второй колонны являются второй компонент азеотропной пары (дистиллат) и регенерированный экстрактивный агент (куб), который направляется на рецикл.
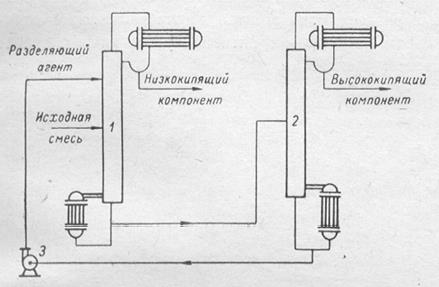
Рис.3 Схема установки для экстрактивной ректификации
1 – экстрактивно-ректификационная колонна; 2 – отгонная колонна
В качестве экстрагентов аренов в промышленности применяются сульфолан, диэтиленгликоль (ДЭГ), триэтиленгликоль (ТЭГ), тетраэтиленгликоль, N-формилморфолин, диметилсульфооксид, смеси N-метилпирролидона или N-метилкапролактама с этиленгликолем. Важнейшие требования к экстрагентам, от которых зависят степень извлечения и качество выделенных аренов, следующие:
· высокая групповая селективность, которую можно характеризовать отношением коэффициентов активности разделяемых групп углеводородов при бесконечном разбавлении в экстрагенте (предельных коэффициентов активности), например отношением предельных коэффициентов активности циклогексана и бензола (γ
0
цг
/γ
0
б
);
· высокая растворяющая способность по отношению к аренам, от которой зависит степень их извлечения и требующееся соотношение экстрагента к сырью; растворяющая способность, или «емкость», экстрагента тем выше, чем ниже предельные коэффициенты активности аренов, поэтому ее можно характеризовать величиной, обратной предельному коэффициенту активности бензола в экстрагенте (1/γ
0
б
);
· низкая селективность по молекулярным массам, которая зависит от отношения предельных коэффициентов активности углеведородов – гомологов, напрмер, октана и гептана (lg(γ
0
окт
/γ
0
гп
)); только при этом условии экстрагенты высокой групповой селективностью могут обеспечить высокую степень извлечения и качество не только бензола, но и его гомологов – толуола, ксилолов.
Характеристика промышленных экстрагентов по всем этим важнейшим критериям представлена в табл.1. Некоторые селективные растворители с повышенной растворяющей способностью – N-метилпирролидон, N-формилморфолин, а также диметилформамид – применяются не только в качестве экстрагентов, но и как растворители для выделения аренов С6
-С8
из более узкокипящих фракций в процессах экстрактивой ректификации.
Зависимость групповой селективности экстрагентов от температуры представлена на рис.4. С повышением температуры селективность сильно ассоциированных растворителей, в частности этиленгликоля, снижается более резко, чем слабо ассоциированных экстрагентов с повышенной растворяющей способностью, к которым относятся N-метилкапролактам, N-метилпирролидон, диметилформамид, N-формилморфолин.
Табл.1
Предельγные коэффициенты активности углеводородов, селективность экстрагентов по отношению к системе циклогексан-бензол (γ0
цг
/γ0
б
), селективность по молекулярным массам (
lg
(γ0
окт
/γ0
гп
)) и растворяющая способность экстрагентов (1/γ0
б
)
| Экстрагент
|
Т,0
С
|
γ0
гп
|
γ0
окт
|
γ0
цг
|
γ0
б
|
γ0
цг
/γ0
б
|
lg
(γ0
окт
/γ0
гп
)
|
(1/γ0
б
)
|
| Сульфолан |
30
60
80
|
99,0
60,0
50,7
|
141
80,1
65,6
|
33,8
23,0
19,9
|
2,43
2,38
2,49
|
13,9
9,66
7,99
|
0,154
0,125
0,122
|
0,412
0,420
0,402
|
| N-формилморфолин |
30
61,7
|
46,68
32,06
|
63,35
41,78
|
17,90
13,77
|
2,03
1,99
|
8,82
6,92
|
0,133
0,115
|
0,493
0,503
|
| Диметилсульфоксид |
20
40
60
|
149
95
65
|
220
136
87
|
46,0
33,0
25,0
|
3,83
3,20
3,03
|
12,0
10,3
8,25
|
0,169
0,159
0,126
|
0,261
0,312
0,330
|
| Этиленгликоль |
20
40,8
60
|
1370
930
457
|
2380
1440
663
|
278
258
188
|
31,6
33,3
32,0
|
8,80
7,75
5,88
|
0,240
0,190
0,162
|
0,032
0,030
0,031
|
| Диэтиленгликоль |
25
60
100
|
164,5
-
-
|
260
-
-
|
71,7
-
-
|
6,41
6,5
6,2
|
11,2
-
-
|
0,199
-
-
|
0,156
-
-
|
| Триэтиленгликоль |
30
80
|
94,5
40,8
|
139
54,5
|
29,3
15,2
|
3,86
3,02
|
7,59
5,03
|
0,168
0,126
|
0,259
0,331
|
| Тетраэтиленгликоль |
30
70
|
57,9
-
|
85,8
-
|
18,3
-
|
2,46
2,48
|
7,44
-
|
0,171
-
|
0,407
0,403
|
| N-метилпирролидон |
30
60
|
17,7
11,5
|
21,6
13,1
|
8,52
6,30
|
1,08
1,08
|
7,89
5,83
|
0,086
0,057
|
0,926
0,926
|
| N-метилкапролактам |
20
40
60
|
7,9
6,8
5,8
|
9,1
7,1
6,7
|
4,2
4,0
3,3
|
0,85
0,85
0,87
|
4,9
4,7
3,8
|
0,061
0,019
0,063
|
1,176
1,176
1,149
|
| Диметилформамид |
25
40
|
22,7
18,9
|
29,4
24,2
|
11,6
9,9
|
1,47
1,43
|
7,89
6,92
|
0,122
0,107
|
0,680
0,699
|
| N-метилморфолинон-3 |
30 |
55,7 |
80,4 |
21,9 |
2,20 |
9,95 |
0,159 |
0,455 |
| N-ацетилоксазолидин |
30 |
40,6 |
52,6 |
17,6 |
1,84 |
9,57 |
0,089 |
0,543 |
| N-метилоксазолидинон-2 |
30 |
53,1 |
70,4 |
21,2 |
1,98 |
10,7 |
0,123 |
0,505 |
| Тиетан-1-оксид |
30 |
59,6 |
79,0 |
21,2 |
1,97 |
10,8 |
0,122 |
0,508 |
| Тиофан-1-оксид |
30 |
38,2 |
53,1 |
14,6 |
1,64 |
8,9 |
0,143 |
0,610 |
| 2-метилтиетан-1,1-диоксид |
30 |
44,2 |
58,8 |
18,1 |
1,90 |
9,53 |
0,124 |
0,526 |
| 2-тетрагидрофурфурил-оксипропионитрил |
30 |
15,6 |
19,6 |
7,10 |
1,17 |
6,07 |
0,099 |
0,855 |
| Цианэтильные производные метилденглицеринов |
30
40
|
77,0
62,8
|
109
88,2
|
24,5
20,8
|
2,26
2,12
|
10,8
9,81
|
0,151
0,147
|
0,442
0,472
|
| Левулинонитрил |
30 |
63,0 |
90,0 |
24,0 |
2,43 |
9,88 |
0,155 |
0,412 |
Рис.4
Зависимость селективности растворителей по отношению к системам гексан-бензол и циклогексан-бензол от температуры
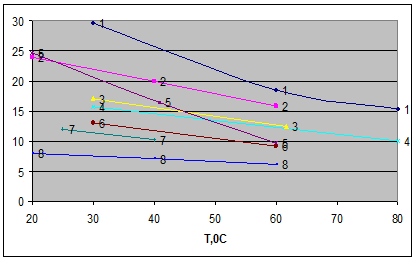
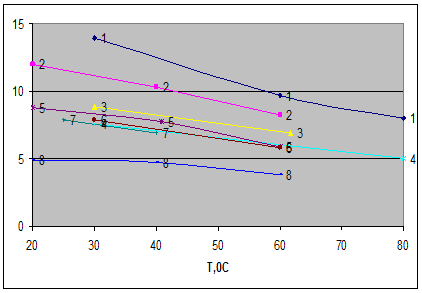
1-сульфолан, 2-диметилсульфоксид, 3 - N-формилморфолин, 4-ТЭГ, 5-этиленгликоль, 6 - N-метилпирролидон, 7 – диметилформамид, 8 - N-метилкапролактам.
По селективности по отношению к системе циклогексан-бензол при 60 0
С экстрагенты располагаются в следующий ряд: сульфолан > диметилсульфооксид > N-формилморфолин > ТЭГ ≈ тетраэтиленгликоль ≈ ДЭГ ≈ ЭГ > N-метилпирролидон диметилформамид > N-метилкапролактам.
Практически в той же последовательности располагаются экстрагенты и по селективности к системе циклогексан – бензол, только в этом случае N-метилпирролидон и диметилформамид оказываются близки к гликолям.
По растворяющей способности к аренам при одинаковой температуре экстрагенты располагаются в следующий ряд: N-метилкапролактам > N-метилпирролидон > диметилформамид > N-формилморфолин ≈ сульфолан > тетраэтиленгликоль > диметилсульфооксид > ТЭГ > ДЭГ > этиленгликоль.
Низкая растворяющая способность характерна для сильно ассоциированных растворителей – гликолей, диметилсульфооксида. Эти же растворители проявляют и повышенную селективность по молекулярным массам, что обусловлено высокими значениями удельных энтальпий образования полости в структуре ассоциированных экстрагентов и быстро возрастающими затратами энергии при растворении углеводородов-гомологов с увеличением их молярных объемов.
В табл. 1 представлены также данные о селективности и растворяющей способности ряда растворителей, предложенных для экстракции аренов в результате многолетних исследований, проводимых в Санкт-Петербурском государственном технологическом институте. По сочетанию высокой групповой селективности и растворяющей способности, умеренной селективности по молекулярным массам эти растворители не уступают наиболее эффективным промышленным экстрагентам – сульфолану и N-формилморфолину.
Предложенные селективные растворители не нашли применения из-за отсутствия их промышленного производства.
К экстрагентам предъявляется еще и ряд технологических требований:
· Плотность, отличающаяся от плотности сырья, - для быстрого расслаивания экстрактной и рафинатной фаз;
· Температура кипения, отличающаяся от температуры кипения компонентов сырья, - для регенерации экстрагента из экстрактной фазы ректификацией;
· Хорошая растворимость в воде и высокие коэффициенты распределения при экстракции водой из рафинатной фазы и экстракта;
· Низкая вязкость, что повышает коэффициент полезного действия тарелок экстракционной колонны или снижает высоту, эквивалентную теоретической ступени экстракции;
· Высокая термическая и гидролитическая стабильность – при температуре в колонне регенерации экстрагента из экстрактной фазы ректификацией с водяным паром;
· Низкая коррозионная активность;
· Невысокая температура плавления;
· Низкая удельная теплоемкость и теплота испарения – для снижения энергозатрат при нагревании и глубокой регенерации экстрагента вакуумной ректификацией;
· Доступность сырья для производства и низкая стоимость экстрагента;
· Низкая токсичность;
· Взрывобезопасность.
Физико-химические свойства экстрагентов, применяющихся в промышленности для выделения аренов С6
-С8
, представлены в табл.2. Преимущества и недостатки применяющихся селективных растворителей сопоставлены в табл.3.
Табл.2
Физико-химические свойства экстрагентов аренов
| Экстрагент |
ρ20
4
|
Ткип
,0
С |
Тпл
,0
С |
η
(при 200
С), мПа*с
|
Ср,( при 20 0
С), кДж/(кг*К) |
Нисп
, (при 25 0
С), кДж/моль |
σ
(при 200
С), мН/м
|
ПДК,
мг/м3
|
| Сульфолан |
1,2604
(300
С)
|
285 |
28,4 |
10,0
(300
С)
|
1,34
(300
С)
|
61,5
(2000
С)
|
60,33
(400
С)
|
50 |
| Этиленгликоль |
1,1135 |
197,6 |
-12,6 |
19,9 |
2,40
(220
С)
|
52,5
(197,60
С)
|
48,43 |
0,1 |
| Диэтиленгликоль |
1,1161 |
245,8 |
-7,8 |
35,7 |
2,093 |
62,0 |
48,5
(250
С)
|
0,2 |
| Триэтиленгликоль |
1,1242 |
285 |
-4,3 |
49,0 |
2,17 |
71,6 |
45,57 |
- |
| Тетраэтиленгликоль |
1,1247 |
327,3 |
-6,2 |
61,3 |
2,14 |
88,8 |
45
(250
С)
|
- |
| N-формилморфолин |
1,1528 |
244 |
20-21 |
9,37 |
1,97 |
46,06 |
- |
- |
| Диметилсульфооксид |
1,0960
(250
С)
|
189 |
18,45 |
2,473 |
2,05 |
57,28 |
43,49 |
20 |
| N-метилпирролидон |
1,0328 |
202 |
-24 |
1,65
(250
С)
|
1,97 |
53,06 |
39,91 |
100 |
| N-метилкапролактам |
1,0129 |
237 |
6,0 |
5,61 |
1,95 |
61,6 |
39,9 |
- |
| Диметилформамид |
0,9445
(250
С)
|
153 |
-61 |
0,80 |
2,05 |
47,4 |
36,76 |
10 |
Табл. 3
Сравнительная характеристика селективных растворителей
| Селективный растворитель |
Преимущества растворителя |
Недостатки растворителя |
| Диэтиленгликоль |
Достаточно высокая Ткип, низкая Ткрист, высокая плотность, относительно низкая стоимость, достаточно высокая стабильность, малая коррозионная активность, полная смешиваемость с водой и высокие коэффициенты распределения ДЭГ при водной отмывке рафинатной фазы и экстракта |
Низкая растворяющая способность по отношению к аренам, невысокая групповая селективность, высокая селективность по молекулярным массам, низкие коэффициенты распределения аренов, высокая вязкость, высокая теплоемкость |
| Триэтиленгликоль |
То же |
То же, но растворяющая способность по отношению к аренам выше, чем у ДЭГ |
| Тетраэтиленгликоль |
То же (кроме Ткип) |
То же, но растворяющая способность выше, чем у ДЭГ и ТЭГ; чрезмерно высокая Ткип, что осложнает регенерацию тетраэтиленгликоля |
| Сульфолан |
Наивысшая групповая селективность по сравнению с другими экстрагентами , высокая плотность, низкая теплоемкость, достаточно высокая стабильность |
Меньшие коэффициенты распределения сульфолана при водной отмывке его рафинатной фазы и экстракта, необходимость вакуумной отгонки аренов из экстрактной фазы, высокая Ткрист
|
| Диметилсульфоксид |
Достаточно высокая групповая селективность(выше, чем у гликолей), низкая вязкость |
Низкая термическая и гидролитическая стабильность, что приводит к необходимости регенерации ДМСО реэкстракцией аренов низкокипящими алканами; невысокая растворяющая способность и коэффициенты распределения аренов. |
| Смесь N-метилпирролидон-этиленгликоль ≈ 60/40 %(масс.) |
Высокая растворяющая способность по отношению к углеводородам, высокие коэффициенты распределения аренов, низкая селективность по молекулярным массам, низкая вязкость, высокая термическая и гидролитическая стабильность, полная смешиваемость с водой, низкая токсичность |
Невысокая групповая селективность по отношению к аренам С6-С8
, высокая стоимость растворителя |
| Смесь N-метилкапролактам –этиленгликоль ≈ 35/65 %(масс.) |
То же, но с введением в N-метилкапролактам этиленгликоля эти преимущества в значительной степени нивелируются |
Низкая групповая селективность ( ниже, чем у N-метилпирролидона) |
| N-формилморфолин |
Высокая групповая селективность и низкая селективность по молекулярным массам, позволяющие выделить бензол и толуол в одной колонне экстрактивной ректификации; достаточно высокая растворяющая способность, что дает возможность использовать растворитель не только при экстракции, но и при экстрактивной ректификации; высокая стабильность |
Высокая Ткрист
|
| Диметилформамид |
Высокая растворяющая способность, низкая вязкость |
Невысокая гидролитическая стабильность; коррозионная активность; токсичность |
Однако все предложенные в последние годы экстрагенты и их смеси уступают по селективности к аренам наиболее эффективным растворителям – сульфолану и N-формилморфолину, применяющимся в промышленности.[4]
2.2.1.
Экстракция аренов С6
-С8
сульфоланом из катализатов риформинга.
Первая публикации о разработке промышленного процесса экстракции аренов С6
-С8
из катализатов риформинга появилась в 1959г. К.Г. Дил с соавторами сообщили о разработке фирмами ShellDevelopment и ShellOil процесса экстракции бензола, толуола и ксилолов, более эффективного по сравнению с Udex – процессом, в котором применялся диэтиленгликоль. Отмечалось, что разработанный процесс может быть использован и для повышения октанового числа моторных топлив.
Капитальные затраты на строительство промышленной установки оценивалась в 75% от капитальных затрат на установку Udex – процесса. В качестве полярного экстрагента предлагался сульфолан с 1.3% (масс) воды при массовом отношению к сырью 6.8 : 1, а в качестве промывного растворителя – высокипящая парафиновая фракция со средней молярной массой 460 (типа гексадекана, но МС16Н34 = 226).
Температура процесса экстракции рекомендовалась 212 0
F, а температура низа колонны отгонки аренов из экстрактной фазы 375 0
F ( 100 и 90 0
C соответственно).
В следующем сообщении тех же авторов отмечаются преимущества сульфолана как экстрагента аренов по сравнению с диэтиленгликолем: более высокая селективность и растворяющая способность по отношению к аренам, более высокая термоокислительная стабильность, меньшая вязкость и теплоемкость. В связи с жэтим удельные энергозатраты при использовании диэтиленгликоля и сульфолана составляют 587 и 206 тыс.ккал/м3
сырья. Однако коэффициенты распределения сульфолана приводной отмывке его из рафинатной фазы и экстракта ниже, чем коэффициенты распределения диэтилегликоля, поэтому для отмывки сульфолана необходима экстракционная колонна эффективностью в несколько теоретических ступеней.
На установке выделения аренов из катализата риформинга в Хьюстане, США, заменили экстрактивную ректификацию с фенолом на экстракцию сульфоланом. Уже на этой установке отказались от использования высококипящих парафинов в качестве промывного растворителя. Насыщенные углеводороды, как и в схеме Udex – процесса остающиеся в экстрактной фазе, отгоняли с острым водяным паром и рисайкл возвращали в экстрактор. Степень извлечения бензола составляет 99.7%, толуола 98.0% и ксилолов 80%, а содержание основного вещества в товарных продуктах 99.96, 99.9 и 99.75% (масс.) соответственно.
Близкие к отмеченным выше результаты были достигнуты и на первой установке экстракции аренов сульфоланом, построенной в 1961 г. в Италии.
Впоследствии показатели работы установок были улучшены, степень извлечения аренов составила, %: бензол – 99.9, толуол – 99.5, арены С8 – 98, а содержание неароматических примесей в бензоле снизилось до 0.01, в толуоле – до 0.02 и аренах С8
– до 0.1% (масс.)
Принципиальная технологическая схема процесса представлена на рис.5. Регенерация сульфолана из экстрактной фазы проводится ректификацией с острым водяным паром. В колонне 2 отпаривается рисайкл (остающиеся в экстрактной фазе насыщенные углеводороды вместе с частью бензола), который возвращается в экстрактор 1. В колонне 3 с острым водяным паром и при небольшом разрежении для снижения температуры низа колонны отгоняется экстракт, который после отделения от воды в сепараторе 6 разделяется ректификацией на товарный бензол, толуол и арены С8. Рафинатная фаза промывается водой в экстракционной колонне 4 для удаления растворенных примесей сульфолана.
Рис.5
Принципиальная технологическая схема процесса экстрактивной ректификации аренов С6
-С8
из фракции 62-140 0
С катализата риформинга
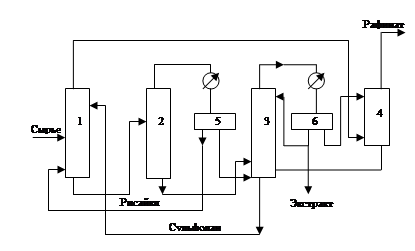
1-экстрактор; 2-ректификационная колонна (отпаривание рисайкла); 3-колонна выделения экстракта; 4 –колонна водной промывки рафинатной фазы; 5,6-сепараторы.
В промышленной эксплуатации к 1992 г. находились 107 установок экстракции аренов сульфоланом. Мощность установок удалось повысить при использовании в экстракторе новых тарелок фирмы UOP типа «MultipleUpcomer», а в отпарных колоннах – «MultipleDowncomer», при этом пропускная способность колонн возросла на 35%.
Д. Григориу с сотрудниками исследовано влияние различных факторов на коэффициенты разделения углеводородов при экстракции аренов сульфоланом (и другими экстрагентами) из фракции 65-154 0
С катализата риформинга (табл.4).
Табл. 4.
Влияние параметров процесса экстракции аренов сульфоланом из фракции 65-154 сульфоланом из фракции 65-154 0
С сульфоланом из фракции 65-154 0
С катализата риформинга на коэффициенты разделения β
| t, 0
C
|
β
|
S\F
|
β
|
Содержание воды,%(масс.)
|
β
|
| 100 |
9.32 |
0.26:1 |
6.26 |
1 |
9.32 |
| 125 |
7.91 |
5.65:1 |
8.69 |
4 |
9.90 |
| 150 |
6.99 |
21.4:1 |
9.90 |
7 |
11.1 |
Влияние температуры процесса на значения β исследовалось при содержании воды в сульфолане 1% (масс.); влияние массового соотношения сульфолан :сырье (S\F) – при 100 0
С, содержание воды в сульфолане 4% (масс.); влияние содержания воды в сульфолане – при 100 0
С и концентрации аренов в сырье 9% (масс.) как следует из табл.4, коэффициенты разделения увеличиваются при снижении температуры, повышения соотношения сульфолан : сырье и добавлении воды к сульфолану. Полученные результаты можно объяснить аналогичным влиянием рассмотренных факторов на групповую селективность, которая хорошо коррелирует с коэффициентами разделения. Отмечено, что влияние соотношения экстрагент : сырье на значение β проявляется для сульфолана более резко, чем для диметилсульфоксида и N-метилпирролидона с 10% (масс.) воды. Добавление же воды, наоборот, в меньшей степени повышает селективность сульфолана, чем селективность (lg β) диметилсульфоксида и N-метилпирролидона.
Различный характер изменения селективности отмечен и при добавлении воды к сульфолану и N-формилморфолину: обводнение сульфолана резко снижает его растворяющую способность по отношению к аренам, мало влияя на селективность; добавление же воды к N-формилморфолину значительно повышает селективность при относительно малом снижении растворяющей способности.
В работах приведены результаты одноступенчатой экстракции аренов С6
-С9
из катализата риформинга сульфоланом и другими экстрагентами при их массовом отношении к сырью 3:1 (табл.5). Как следует из табл.5, сульфолан уступает по основным результатам процесса экстракции диметилсульфоксиду, N-метилпирролидону и диметилформамиду с 10%(масс.) воды.
Однако сопоставление эффективности различных экстрагентов проведено не в оптимальных условиях: так, диметилсульфоксид применятся на промышленных установках при содержании воды 9% (масс.), N-метилпирролидон – при температуре 50-60 0
С, а диметилформамид имеет невысокую гидролитическую стабильность и поэтому используется в безводном состоянии в процессе экстрактивной ректификации, а не экстракции.
Таблица 5.
Результаты одноступенчатой экстракции аренов С6
-С9
из катализата риформинга
.
| Экстрагент |
Содержание воды в экстрагенте, %(масс.) |
t, 0
C |
Содержание аренов, %(масс.) |
Степень извлечения аренов, % |
β |
| сырье |
экстракт |
| Сульфолан |
1.0 |
100 |
28.0 |
60.0 |
56.5 |
7.8 |
| Диметилсульфоксид |
0.05 |
50 |
30.0 |
67.5 |
64.0 |
11.3 |
| Диоксид серы |
- |
-30 |
24.5 |
49.0 |
80.6 |
12.0 |
| N-метилпирролидон |
10.0 |
25 |
29.5 |
67.5 |
63.4 |
10.5 |
| Этиленкарбонат |
0.3 |
90 |
30.0 |
73.0 |
44.5 |
11.7 |
| Морфолин |
14.0 |
25 |
28.5 |
67.5 |
35.6 |
7.25 |
| Фурфурол |
4.0 |
25 |
30.0 |
52.5 |
67.3 |
5.28 |
| Диметилформамид |
10.0 |
25 |
30.0 |
63.5 |
60.5 |
9.1 |
| Тиодипропионитрил |
- |
65 |
28.8 |
82.0 |
48.4 |
19 |
| Оксидипропионитрил |
- |
65 |
28.0 |
71.5 |
51.5 |
11.5 |
| Диэтилгликоль |
10.0 |
150 |
29.5 |
71.0 |
24.0 |
7.3 |
В то же время сульфолан обеспечивает значительно более высокую степень извлечения аренов по сравнению с диэтиленгликолем в соответствии с более высокой растворяющей способностью сульфолана. Коэффициент разделения углеводородов при использовании сульфолана β = 7.8 представляется заниженным, не соответствующим максимальной селективности по сравнению с другими экстрагентами.
А.З. Биккуловым с сотрудниками проведена оптимизация параметров процесса экстракции аренов сульфоланом из фракций катализата риформинга 62-105 0
С и 105-140 и 105-140 0
С. Оптимальны следуюшие условия:
Для фракции 62-105 0
С – содержание воды в сульфолане 3.22% (об.), объемное соотношение экстрагент : сырье 2.75 : 1, температура 52 0
С, расход рисайкла 51.3% (об.) от сырья, число ступеней контакта 7;
Для фракции 105-140 0
С – содержание воды в сульфолане 8.0% (об.), объемное соотношение экстрагент : сырье около 2.8 : 1, температура 119 0
С, расход рисайкла 44.6% (об.) от сырья, число ступеней контакта 11.2.
Найденные оптимальные режимы экстракции приблизительно соответствуют условиям промышленных процессов, однако содержание воды в сульфолане, в особенности при экстракции ксилолов, представляется завышенным.
В работе исследован процесс экстракции бензола, толуолов и ксилолов сульфоланом в 10-ступенчатом каскаде экстракторов: влияние температуры, соотношения экстрагент : сырье, скорости перемешивания. В качестве оптимальных условий рекомендовано соотношение сульфолан : сырье 3:1 и температура 30 0
С.
Технико-экономические показатели процессов экстракции аренов из различных фракций катализата риформинга сульфоланом и гликолями сопоставлены в табл.6.
Табл. 6
.
Технико-экономические показатели процессов экстракции аренов из фракции 62-140 0
С катализата риформинга сульфоланом и гликолями.
| Показатели |
Сульфолан |
Диэтиленгликоль |
Триэтиленгликоль |
Тетраэтиленгликоль |
| Массовое соотношение экстрагент:сырье |
4:1 |
15:1 |
10:1 |
6:1 |
| Содержание воды в экстрагенте, % (масс.) |
<1.5 |
7 |
5 |
5 |
| Расход рисайкла %(масс.)на сырье |
50 |
80 |
80 |
80 |
| Температура, 0
С |
100 |
150 |
150 |
150 |
| Давление в экстракторе, МПа |
0.5 |
0.8-1 |
0.8-1 |
0.8-1 |
| Расход на 1 т аренов: |
| пар, Гкал |
1.99 |
2.7 |
2.45 |
2.21 |
| Электроэнергия, кВт*ч |
61.1 |
71.5 |
61.7 |
50.0 |
| Вода оборотная,м3
|
6.3 |
10.1 |
9.7 |
9.3 |
Себестоимость производства аренов экстракцией сульфоланом ниже не только по сравнению с экстракцией гликолями, но и другими экстрагентами. Так, она составляет 89.4% от себестоимости аренов, выделенных с использованием смешанного экстрагента N-метилпирролидон – этиленгликоль.
Сульфолан наиболее эффективный экстрагент аренов из нафты – фракции с концом кипения до 200 0
С.
С использованием метода UNIFACв работе рассчитаны равновесные составы фаз в процессе экстракции бензола из смеси с гексаном, селективность и емкость (растворяющая способность) 21 полярного растворителя, а также энергозатраты при экстракции аренов и регенерации экстрагентов. Наиболее селективным и наилучшим растворителем с учетом критерия минимальных энергетических затрат оказался сульфолан.
Метод UNIFAC был использован и для моделирования фазовых равновесий жидкость – жидкость в системе сульфолан – арены С6
-С8
– алканы С6
-С8
. Рассчитанные составы равновесных экстрактной и рафинатной фаз удовлетворительно согласуются с экспериментальными данными.
Экологическими требованиями во многих странах мира ограничивается содержание аренов, и прежде всего бензола, в бензине. Так, в Японии к 1999г. допустимое содержание бензола в бензине должно быть снижено до 1% (масс.), для чего рекомендуется экстракция сульфоланом. Многие нефтеперерабатывающие компании (NipponOil, CosmoOil, ShowaShell, KyokutoOil, KoaOil) уже начали строительство установок экстракции бензола из бензина.
Ряд патентов относится к методам регенерации сульфолана из экстрактной и рафинатной фаз в процессе экстракции аренов. Так, вместо острого водяного пара в качестве отпаривающего агента предлагается бутан. Из рафинатной фазы сульфолан рекомендуется адсорбировать на активированном угле, а в качестве десорбентов использовать арены. Удаление сульфолана из экстрактной и рафинатной фаз возможно также адсорбцией силиагелем или активированным оксидом алюминия с последующей десорбцией сульфолана сырьем – катализатом риформинга.
Регенерировать сульфолан предложено диффузией через мембраны. Однако все эти методы регенерации сульфолана не нашли пока промышленного применения.
Из экстрактной фазы сульфолан регенерируют отпариванием углеводородов с острым водяным паром, а из рафинатной фазы – реэкстракцией водой. Для более полного удаления сульфолана предлагается повышать линейную скорость промывной воды: так, при промывке рафинатной фазы, содержащей 2.7% (мол.) аренов и 1.2% (мол.) сульфолана, с линейной скоростью воды 0.3 м/с р рафинате остается 50-100 ppm сульфолана, а при скорости 1.85 м/с -10-12 ppm. Можно увеличить соотношение воды и рафинатной фазы: рафинатьпропускать с линейной скоростью 1.5 -2.4, в то время как для реэкстракции полиалкиленгликолей достаточно молярное соотношение 0.5-1. Возможно применение двухстадийной водной отмывки сульфолана из рафинатной фазы.
Реконструкция блока регенерации сульофлана на заводе в Токуяма (Япония) позволила сократить продолжительность операций по новой технологии на 38.3%.[5]
2.3. Процесс обратимой ректификации, как процесс обладающий минимальной энтропией
.
При обычной адиабатической ректификации в колонах конечной протяженности в каждом сечении имеет место неравновесность между паром, поднимающимся с нижележащей ступени контакта, и жидкостью, стекающей с вышележащей ступени. В любом сечении колонн с дифференциальным изменением состава фаз по высоте (пленочные, насадочные) в принятых условиях также наблюдается неравновесность.
В адиабатических ректификационных колоннах бесконечной эффективности как дискретного типа изменения состава фаз, так и непрерывного термодинамическое равновесие достигается только в зонах постоянных концентраций, где процесс ректификации становится обратимым.
Если равновесие фаз имеет место в каждом сечении колонны любого типа, то осуществляется термодинамически обратимый процесс ректификации. Такой процесс характеризуется следующими требованиями:
1. бесконечным числом ступеней разделения, соответствующим каждому составу, лежащему на траектории ректификации;
2. бесконечно малой скоростью изменения контактирующих фаз и их количеств;
3. дифференциальным подводом и отводом тепла по высоте ректификационной колонны;
4. наличием на каждой ступени разделения n-2 распределенных компонентов между кубом и дистиллятом, где n – число компонентов, поступающих на данную стадию.
В процессе ректификации происходит, с одной стороны, изменение энтропии потоков внутри колонны, а с другой стороны, изменение энтропии источника (в кипятильнике) и приемника (в дефлегматоре) тепла. Энтропия потоков внутри ректификационной колонны уменьшается (энтропия продуктов меньше энтропии сырья). Энтропия источника и приемника тепла в сумме увеличивается за счет передачи тепла от источника с высокой температурой к приемнику с низкой температурой. Во всех реальных процессах ректификации увеличение энтропии за счет передачи тепла значительно больше, чем ее уменьшение в самом процессе ректификации за счет процесса разделения. Таким образом, в целом происходит увеличение энтропии, связанное с различными источниками термодинамических потерь (неравномерность на тарелках, смешение потоков в питании и на концах колонны, гидравлические сопротивления, температурные напоры в теплообменниках и т. д.).
Для термодинамически обратимого процесса ректификации суммарное изменение энтропии в самой ректификационной колонне, в источниках и в приемниках тепла должно быть равно нулю [1].
2.4. Способы приближения необратимых процессов ректификации к процессу обратимой
ректификации.
В силу своих особенностей процесс обратимой ректификации обладает наименьшими энергозатратами, однако не может быть осуществлен на практике. Из приведенных в предыдущем разделе требований для реализации процесса обратимой ректификации на практике полностью может быть осуществлено лишь четвертое. Поэтому в реальных процессах может быть достигнуто лишь приближение к термодинамической обратимости за счет определенных структурных особенностей схемы или усовершенствования массообменных устройств.
Одним из способов приближения реальных процессов ректификации к термодинамически обратимым является применение колонн со стриппинг-секциями, являющихся примером колонн с частично связанными тепловыми и материальными потоками. Колонна со стриппингом представляет собой сложную колонну с дополнительной боковой отпарной или укрепляющей секцией, снабженной кипятильником или конденсатором соответственно. В стриппинг-секцию подается поток бокового погона, одна часть которого отбирается в качестве продукта, а другая возвращается обратно в главную колонну (см. рис.6). На одной колонне может устанавливаться несколько стриппингов. Колонны со стриппингами более сложны в управлении по сравнению с простыми колоннами, однако позволяют существенно снизить энергозатраты, в связи с чем они получили широкое применения в химической технологии, особенно в процессах переработки нефти.
Рис.6.
Сложные колонны с боковой секцией: а – отпарной, б – укрепляющей.
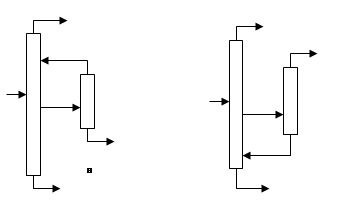
2.4.2. Многокорпусная ректификация.
Другой способ приближения необратимого процесса ректификации к обратимому – многокорпусная ректификация. Схема установки многокорпусной ректификации изображена на рис.7. Установка состоит из последовательности простых дистилляционных колонн. Поток питания F распределен между колоннами в установке. Особенность процесса заключается в том, что дистиллят предыдущей колонны используется для подогрева куба последующей. Для этого необходимо выполнение условия:
tD
1
> tW
2
,
tD
2
> tW
3
; (2.3.2.1)
где tD
1
и tD
2
температура дистиллята 1-й и 2-й колонны соответственно, а tW
2
и tW
3
температура куба 2-й и 3-й колонны. Это условие выполняют поддержанием в колоннах разного давления, т. е. P1
> P2
> P3
, где P1
, P2
, P3
– рабочие давления 1-й, 2-й и 3-й колонн соответственно, причем для удобства эксплуатации и управления, давления стараются подбирать так, чтобы давление последней колонны в последовательности было атмосферным. Установки многокорпусной ректификации требуют существенных капиталовложений и имеют ограничения в применении требованием относительно близких температур кипения и термической устойчивости компонентов разделяемой смеси.
Рис. 7.
Разделение бинарной смеси
AB
в схеме многокорпусной ректификации.
Одним из наиболее перспективных методов приближения процесса необратимой ректификации к обратимой, является теплоинтеграция потоков. В системах колонн, связанных материальными и тепловыми потоками предыдущая и последующая колонны связаны противоположно направленными паровыми и жидкостными потоками, соединяющими верх и низ предыдущей колонны с точками ввода питания последующей колонны или верх последующей колонны с боковым отбором предыдущей и, наконец, низ последующей колонны с боковым отбором предыдущей.
Применение системы ректификационных колонн, связанных материальными и тепловыми потоками, позволяет на 20 – 50 % снизить общие затраты тепла и холода по сравнению с простыми колоннами. Независимо от числа получаемых продуктов технологические схемы установок, где используются колонны, связанные материальными и тепловыми потоками, имеют один дефлегматор и один кипятильник, однако на практике такие схемы колонн эффективны только при разделении близкокипящих многокомпонентных смесей, так как лишь в этом случае в последней колонне могут быть приняты температуры верха и низа, удовлетворяющие экономичным условиям конденсации и испарениям получаемых продуктов [7]. На рисунке 8 изображен пример колонн, с полностью связанными материальными и тепловыми потоками.
Рис. 8.
Ректификационные колонны с полностью связанными материальными потоками.
Альтернативный способ приближения необратимого процесса ректификации к обратимому путем структурных изменений схем ректификации - усовершенствование контактных устройств ректификационных колонн с целью увеличения их КПД. В химической технологии широко используются как насадочные, так и тарельчатые ректификационные колонны. Уже создано и запатентовано большое количество различных типов насадок и тарелок. Основными факторами, влияющими на выбор контактного устройства для конкретного процесса являются:
· Производительность установки,
· Перепад давления,
· Эффективность контактного устройства
· Коррозия рабочей среды
· Стоимость контактного устройства
При использовании одного и того же типа контактного устройства для разделения различных смесей, оно будет показывать разную эффективность. Это означает, что для каждого конкретного технологического процесса следует подбирать свой, наиболее эффективный в данном случае, тип контактного устройства. Равновесную тарелку, обладающую КПД = 100% создать невозможно, однако ведутся работы по созданию контактных устройств с КПД, приближающимся к 100%. Из известных на сегодняшний контактных устройств, самым высоким КПД обладает провальная тарелка Зульцера с неподвижными клапанами (КПД = 85% для смеси этан-пропан-пропилен).
2.5. Методы воздействия на эффективность работы ректификационных колонн.
В процессе ректификации одной из важнейших технологических задач является воздействие на его протекание с целью получения продуктов необходимой чистоты.
В ходе проектирования ректификационной колонны такое воздействие возможно путем увеличения числа тарелок или высоты слоя насадки, а также использования наиболее эффективных конструкций тарелки или типа и размера элементов насадки.
В ходе эксплуатации ректификационной колонны повысить чистоту продуктов можно, повышая флегмовое число R
:
-увеличением потока флегмы (при сохранении отборов дистиллята и кубового остатка), если кипятильник и конденсатор рассчитаны с некоторым запасом и позволяют повысить тепловую нагрузку;
-уменьшением производительности колонны по исходной смеси, а значит, и по продуктам; при этом, сохраняя поток флегмы и понижая дистиллят, получают более высокое R.
Повысить флегмовое число можно также путем захолаживания флегмы, т. е. возвращения ее в колонну при температуре ниже температуры дистиллята: холодная флегма будет нагреваться в верхних зонах колонны до температуры кипения за счет конденсации части парового потока, и поток флегмы по колонне возрастает.
Увеличивая число тарелок n (высоту слоя насадки), а также флегмовое число, можно в принципе получить сколь угодно чистый дистиллят и кубовый остаток, но абсолютно чистые НКК и ВКК методом ректификации (и вообще – массообмена) получить невозможно. Ведь точки чистых компонентов располагаются в правом верхнем и левом нижнем углах диаграммы y-x, где движущая сила равна нулю; это означает необходимость безграничного увеличения числа тарелок даже при бесконечном R (что, кстати, тоже невозможно при отборе продуктов).
Еще одна возможность воздействия на чистоту продуктов – смещение точки подачи исходной смеси по высоте колонны. Так, если подать исходную смесь в колонну пониже, то увеличится протяженность укрепляющей части, и дистиллят станет чище; при этом уменьшится протяженность отгонной части колонны, так что кубовый остаток будет больше загрязнен низкокипящим компонентом. Изменение точки питания ректификационной колонны может оказаться полезным в двух случаях:
только один из продуктов должен быть весьма чистым (во втором допускается заметное содержание примеси); тогда следует увеличивать протяженность той части колонны, на выходе из которой нужно получать чистый продукт;
по какой-то причине изменился состав исходной бинарной смеси. Например: содержание НКК в исходной смеси понизилось; для сохранения прежней чистоты дистиллята теперь в укрепляющей части колонны потребуется большее число тарелок (или большая высота слоя насадки), чем раньше, а для сохранения чистоты кубового остатка – меньшее число тарелок в отгонной части колонны. Поэтому исходную смесь нового состава следует подавать в колонну на более низкую тарелку. Конкретно: ее надо подавать в колонну на более низкую тарелку. Конкретно: ее надо подавать в то сечение колонны, в котором этот состав равен составу исходной смеси [8].
При разделении многокомпонентных смесей, так же как и в случае с бинарными смесями, процесс ректификации может проводиться как непрерывно, так и периодически.
Периодическая ректификация осуществляется в одной ректификационной колонне путем последовательного (во времени) получения в виде дистиллята сначала наиболее летучего компонента смеси, а затем – компонентов с более высокими температурами кипения. Компонент смеси с самой высокой температурой кипения остается в кубе колонны в виде кубового остатка. Разумеется, реально получают не отдельные компоненты, а фракции с преимущественным их содержанием. Хотя этот процесс более дешев, по сравнению с непрерывной ректификацией, им пользуются реже, так как он не дает возможности четко разделить компоненты и чрезвычайно сложен в управлении из-за постоянно изменяющихся параметров в ходе работы установки.
Непрерывная ректификация многокомпонентных смесей осуществляется в установках, состоящих из ряда ректификационных колонн непрерывного действия, соединение которых в общую схему может быть различным. Каждая простая колонна разделяет поступающую в нее смесь на два продукта, один из которых дистиллят, а второй – кубовый остаток. Поэтому при наличии хотя бы трех компонентов в исходной смеси их разделение на три продукта в одной простой колонне невозможно. В этом случае нужны две колонны [9]
.
2.7. Поливариантность технологических схем.
Создание оптимальной технологической схемы ректификационного разделения многокомпонентных смесей представляет собой достаточно сложную задачу из-за поливариантности организации процесса. С ростом числа компонентов в смеси резко возрастает число возможных вариантов ее разделения. Общее число вариантов ТСР многокомпонентных зеотропных смесей (Z) при использовании простых двухсекционных колонн определяется выражением:
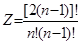 2.7.1 2.7.1
Так, для разделения смеси, состоящей из 3-х компонентов, возможно два различных варианта схемы ректификации, из 5-ти - компонентов - 14, а из 7-ми компонентов - 132.
При использовании М различных методов разделения, таких как обычная и экстрактивная ректификация, абсорбция, экстракция и т.п. Число вариантов ТСР определяется как:
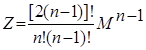 2.7.2 2.7.2
Выбор оптимальных ТСР представляет собой процедуру, которая может быть разбита на два этапа. На первом синтезируются все возможные варианты, а на втором проводится дискриминация по тому или иному критерию. В качестве критерия оптимизации, как правило, выбираются минимальные суммарные приведенные затраты на разделение для всей схемы в целом. Для предварительных оценок применяют другие критерии, например энергозатраты.
Для синтеза технологических схем разделения необходимо знать:
Физико-химические и химические свойства как чистых компонентов, так и всех смесей, составляющих данную многокомпонентную смесь. В том числе температуры кипения компонентов и смесей, параметры фазового равновесия жидкость - жидкость, жидкость - газ, жидкость - пар, жидкость - твердое тело, а также химическую активность компонентов и их термическую стойкость в процессе разделения. Эти свойства позволяют выявить все термодинамические, химические и технологические ограничения, которые необходимо учитывать при синтезе технологических схем разделения.
Возможности различных методов разделения, области их использования, преимущества и недостатки.
Конструктивные особенности и возможности применения различных разделительных аппаратов, располагать классификации таких аппаратов с описанием их структурных характеристик.
Структуру технологических комплексов различного функционального действия, состоящих из ряда аппаратов и применяемых для разделения смесей, обладающих определенными специфическими свойствами. Эти комплексы позволяют преодолеть различные технологические ограничения, связанные с азеотропией, и получить продукты нужного состава.
Методы синтеза технологических схем разделения. С этой целью должны быть рассмотрены как безмашинные методы, так и основанные на применении ЭВМ. В последнем случае необходимо располагать математическими моделями как отдельных элементов и комплексов, так и системы в целом.
Методы оптимизации технологических схем разделения. Оптимизацию технологической схемы необходимо начинать с рассмотрения структуры диаграммы фазового равновесия разделяемой смеси, которая определяет термодинамические ограничения, связанные с азеотропией, и, следовательно, последовательность выделения компонентов или фракций [10,11].
2.8. Термодинамические методы расчета равновесия жидкость-пар.
Выбор модели, адекватно описывающей фазовое равновесие системы, является важным и необходимым шагом при решении массообменного процесса. На сегодняшний день разработано достаточно большое число методов математического моделирования различных типов парожидкостного равновесия.
· Модели локальных составов (Wilson, NRTL, UNIQUAC);
· Уравнения состояния (SRK, PengRobinson);
· Групповая модель (UNIFAC).
Рассмотрим их более подробно.
2.8.1. Модели локальных составов [12].
Концепция локальных составов позволяет учитывать структуру раствора, свойства чистых веществ и межмолекулярные взаимодействия разных типов (слабые неспецифические и сильные специфические). Согласно этой теории раствор рассматривается как упорядоченная структура. Для бинарной смеси можно выделить молекулы двух сортов, при этом молекула одного вида находится в окружении молекул другого вида. Зависимость между концентрациями компонентов внутри такого образования с общей молярной концентрацией компонентов в растворе описывается соотношением, учитывающим вероятность возникновения связей между разноименными молекулами.
Уравнение Вильсона было первым уравнением, в котором была применена концепция локального состава. Основная идея ее состоит в том, что из-за разницы в межмолекулярных взаимодействиях локальный состав вблизи конкретной молекулы в растворе будет отличаться от состава жидкости. Для бинарной пары два параметра связаны со степенью, в которой каждая молекула влияет на состав своего локального окружения. Выражение для коэффициента активности представлено ниже:
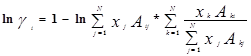
где :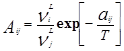 , (a
ij
, °K); , (a
ij
, °K);
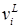 – мольный объем жидкости компонента i. – мольный объем жидкости компонента i.
Параметры aij представляет энергию взаимодействия между молекулами i и j.
Уравнение Вильсона может быть использовано в довольно широком интервале температур, несмотря на то, что его параметры в явном виде не включают температурную зависимость. Эта модель также дает хорошие результаты для смесей, содержащих полярные компоненты.
К недостаткам модели можно отнести то, что уравнение Вильсона не может описывать локальные максимумы или минимумы коэффициента активности, а также не подходит для описания равновесия с частично смешивающимися жидкими фазами.
Уравнение NRTL (non-random two-liquid – неслучайное двужидкостное) было разработано Реноном и Праузницем с целью использовать концепцию локального состава в тех случаях, когда уравнение Вильсона неспособно к предсказанию разделения фаз жидкость – жидкость. Модель NRTL дает хорошие результаты для широкого круга систем, в частности для смесей в высокой степени неидеальных и для частично несмешивающихся систем.
Выражение для коэффициента активности имеет вид:
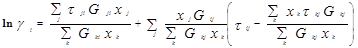
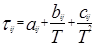 , (когда единицей измерения является °K) , (когда единицей измерения является °K)
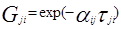
Для каждой бинарной пары требуется три параметра (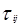 , , 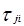 , , 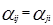 ), которые могут быть расширены включением в них температурной зависимости. ), которые могут быть расширены включением в них температурной зависимости.
Уравнение UNIQUAC (universal quasi-chemical – универсальное квазихимическое) было развито Абрамсом и Праусницем на основании статистически-механических положений и решеточной квазихимической модели Гуггенхайма. Каждую молекулу характеризует два параметра: объем 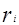 и площадь (поверхность) и площадь (поверхность) 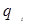 . .
Избыточная энергия Гиббса (и соответственно логарифм коэффициента активности) делится на комбинаторную и остаточную части. Комбинаторная часть зависит только от размеров и форм отдельных молекул, она не содержит бинарных параметров. Остаточная часть, которая учитывает энергетические взаимодействия, имеет два регулируемых бинарных параметра. Выражение для коэффициента активности имеет следующий вид:
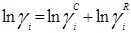
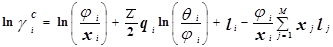
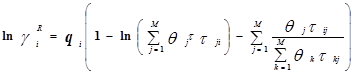
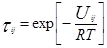 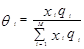
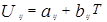 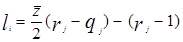
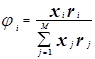 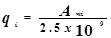 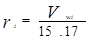 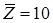
где 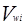 , , 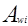 – объем и площадь Ван-дер-Ваальса молекулы i – объем и площадь Ван-дер-Ваальса молекулы i
С
и R
– комбинаторный и остаточный вклады в коэффициент активности жидкости.
Для каждой бинарной пары необходимы два параметра: Uij
и Uji
, которые используются в вычислениях остаточной части коэффициента активности. По желанию параметры могут быть расширены включением температурной зависимости.
Уравнение UNIQUAC адекватно описывает широкий ряд систем, оно применяется для неэлектролитических смесей, содержащих полярные или неполярные компоненты, также подходит для частично смешивающихся систем.
2.8.2 Уравнения состояния[12].
Уравнения состояния применимы для широкого диапазона температур и давлений. Они могут быть использованы для расчетов всех термодинамических свойств, таких как К-значения, энтальпия, энтропия и плотность. Стандартным состоянием как для жидкости, так и для пара является идеальный газ, а отклонения от идеального поведения определяются расчетом коэффициентов фугитивности для обеих фаз.
В 1972 г. для улучшения предсказания парового давления чистых компонентов и парожидкостного равновесия многокомпонентных смесей Соав предложил следующую температурную зависимость:
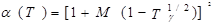
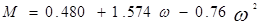
где 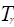 – приведенная температура, Т/Тс – приведенная температура, Т/Тс
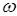 – ацентрический фактор – ацентрический фактор
Константы в уравненииях были получены из преобразования данных по паровому давлению для ограниченного числа обычных углеводородов. Эти пределы использования уравнения состояния СРК ограничены неполярными компонентами.
Уравнение состояния Соав-Редлих-Квонга – это модификация уравнения состояния Редлиха-Квонга (которое основано на уравнении Ван-дер-Ваальса). Соав заменил член 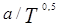 более общей температурной зависимостью а(Т).
Выражение получило следующий вид: более общей температурной зависимостью а(Т).
Выражение получило следующий вид:
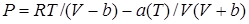
где 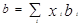 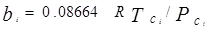
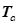 , , 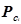 – критические температура и давление для i-го компонента – критические температура и давление для i-го компонента
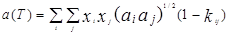
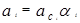 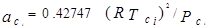
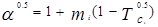
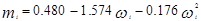
 – ацентрический фактор для компонента i – ацентрический фактор для компонента i
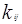 – константа бинарного взаимодействия для компонентов i и j – константа бинарного взаимодействия для компонентов i и j
Введение члена альфа позволило улучшить предсказание парового давления для чистых компонентов. Комбинированная формула для вычисления α(Т) с введенным членом 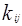 означает улучшение предсказания свойств смеси. означает улучшение предсказания свойств смеси.
Использование формулировки Соава для предсказания свойств смеси включает в себя два этапа. Во-первых, для каждого из компонентов подбирается ацентрический фактор компонента (wi) таким образом, чтобы точно предсказатьдавление паров компонента.
Во-вторых, из экспериментальных данных для бинарных систем с компонентами i и j, для которых достигается фазовое равновесие, определяется параметр kij.
Уравнение состояния Пенга-Робинсона было опубликовано в 1976 году и является модификацией уравнения Редлиха-Квонга. Во многих отношениях оно похоже на уравненеие SRK, но все же он разработано для улучшения предсказания плотности жидкости
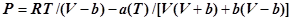
Введением дополнительных членов было достигнуто улучшение предсказания давления пара чистых компонентов и улучшение предсказания свойств смесей.
Использование уравнения Пенга-Робинсона включает в себя два шага, аналогичные предыдущей модели.
2.8.3 Групповые модели[12].
Групповые модели основаны на предположении об аддитивности вкладов различных химических групп в термодинамические свойства компонентов и смесей. Наибольшее распространение получила модель UNIFAC. Именно её мы использовали для создания псевдоэкспериментальных данных парожидкостного равновесия. Рассмотрим модель более подробно.
Метод UNIFAC (universalfunctionalactivitycoefficient – универсальный функциональный коэффициент активности) был разработан в 1975 году Фреденслундом, Джонсом и Праузницем. Данный метод рассчитывает величины коэффициентов активности на основе концепции группового вклада. Предполагается, что взаимодействия между двумя молекулами являются функцией взаимодействий между группами. Данные межгруппового взаимодействия получаются путем обработки экспериментальных данных для пар компонентов. Число функциональных групп ограничено.
Метод UNIFAC основан на модели UNIQUAC, которая представляет избыточную энергию Гиббса (и логарифм коэффициента активности) как комбинацию двух эффектов. Таким образом, используется уравнение:
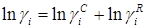
Комбинационный член 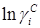 рассчитывается непосредственно по уравнению UNIQUAC с использованием параметров площади и объема Ван дер Ваальса, рассчитанных по индивидуальным структурным группам : рассчитывается непосредственно по уравнению UNIQUAC с использованием параметров площади и объема Ван дер Ваальса, рассчитанных по индивидуальным структурным группам :
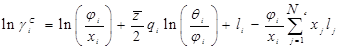
где:
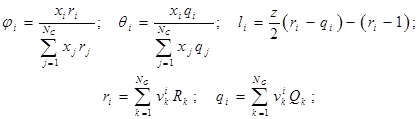
NC
– число компонентов;
NG
– число различных групп в смеси;
z
– согласованное число для пространственной решетки, равное 10;
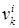 – число функциональных групп типа k
в молекуле i
; – число функциональных групп типа k
в молекуле i
;
Rk
– параметр объема для функциональной группы k
;
Qk
– параметр площади для функциональной группы k
;
xi
– мольная доля компонента i
в жидкой фазе.
Целью настоящей работы является разработка энергосберегающей схемы разделения трехкомпонентной азеотропной смеси бензол – циклогексан - гексан методом экстрактивной ректификации с сульфоланом.
В рамках настоящей работы требуется:
1. Выбрать адекватную модель описания парожидкостного равновесия.
2.Синтезировать все возможные варианты схем разделения трехкомпонентной азеотропной смеси в комплексах простых двухсекционных колонн и эквивалентных им схем с частично связанными тепловыми и материальными потоками.
3. Провести анализ работоспособности синтезированных схем разделения.
4. Произвести расчет и оптимизацию рабочих параметров схем-прообразов состоящих из простых двухсекционных колон и схем-образов содержащих сложные колонны.
5. Среди множества рассмотренных вариантов выявить наиболее экономичную схему исходя из критерия минимальных энергозатрат на разделение
В соответствии с задачами настоящего исследования и для достижения поставленных целей требуется проведение расчетного определения энергетической и экономической эффективности технологических схем ректификации. Для этого использован расчетный эксперимент, базирующийся на математическом моделировании схем ректификации. Достоверность полученных результатов обеспечивается использованием адекватной модели парожидкостного равновесия, строгих моделей и алгоритмов расчета процесса ректификации.
4.1 Программный комплекс PRO/II.
В настоящей работе использован лицензионный программный комплекс PRO/IIwithPROVISION компании SIMSCIcorp., обеспечивающий моделирование и расчет технологических схем ректификации.
PRO/II – компьютерный комплекс для инженерных расчетов процессов органического синтеза и нефтехимии, технологии полимеров и др. Он объединяет базы данных по свойствам химических компонентов и методы расчета термодинамических свойств с гибкими методами расчета аппаратов. Программа обладает вычислительными средствами для выполнения расчетов всех материальных и энергетических балансов, необходимых для моделирования большинства статических процессов. Экспертные системы, расширенная обработка входных данных и проверка ошибок обеспечивают его высокую эффективность и надежность.
Комплекс PRO/II включает базу данных по свойствам более 1750 веществ. Программный комплекс предоставляет пользователю широкие возможности модернизации, как свойств индивидуальных веществ, так и методов расчета, а также позволяет добавлять собственные вещества и аппараты с заданными свойствами.
Ряд программных модулей PRO/II предназначен для расчета процессов ректификации. Расчет ректификационной колонны возможен в проектно – поверочном и поверочном вариантах. В обоих случаях необходимо задать количество тарелок в секциях колонны и состав питания.
Далее представлено описание основных алгоритмов расчета и моделей аппаратов, реализованных в комплексе PRO/II и непосредственно использующихся при расчете схем ректификации.
4.1.1. Методы расчета ректификации.
Все алгоритмы ректификации в программе PRO/II представляют собой строгие модели равновесных ступеней контакта. В каждой модели решаются тепловой и материальный балансы и уравнения равновесия жидкость – пар.
Программа PRO/II предлагает четыре различных алгоритма моделирования ректификационных колонн:
- алгоритм Inside/Out (I/O),
- алгоритм Sure,
- алгоритм Chemdist и
- алгоритм ELDIST.
Алгоритм Chemdist может быть использован для решения большинства задач нефтепереработки и обладает высоким быстродействием. В настоящей работе расчет колонн ректификации проводился по этому алгоритму.
Алгоритм Chemdist представляет собой новый алгоритм, разработанный фирмой SimSci для моделирования в высокой степени неидеальных химических систем. Этот алгоритм представляет собой полный метод Ньютона – Рафсона с полным набором аналитических производных. Он включает в себя производные по составу для коэффициентов фугитивности и активности, позволяет также рассчитать равновесие жидкость – жидкость – пар на любой ступени контакта колонны и поддерживает широкий диапазон конфигураций конденсаторов с двумя жидкими фазами.
Алгоритм Chemdist с химическими реакциями позволяет использовать встроенные процедуры для кинетических реакций, не подчиняющихся закону действующих масс.
Алгоритм Chemdist в программе PRO/II представляет собой метод, основанный на методе Ньютона, и предназначен для решения задач ректификации неидеальных смесей с большим числом (от 10 до 100) химических веществ. Алгоритм позволяет учесть равновесие жидкость – пар, равновесие жидкость – жидкость – пар, а также химические реакции.
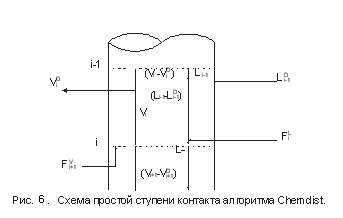
На рис.9 схематически показана равновесная ступень контакта для случая ректификации с двумя фазами и отсутствием химических реакций.
Уравнения, описывающие внутренние тарелки колонны, выглядят следующим образом:
Материальный баланс по компоненту:
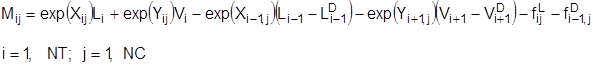 Тепловой баланс: Тепловой баланс:
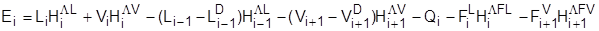 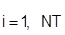
Равновесие жидкость – пар:
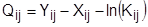
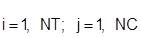
где: Fi
– общий поток сырья на тарелку i;
Li
– общий поток жидкости с тарелки i;
Vi
– общий поток паров с тарелки i;
Qi
– подвод тепла к тарелке i;
Ti
– температура на тарелке i;
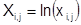 - натуральный логарифм мольных долей жидкости; - натуральный логарифм мольных долей жидкости;
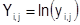 - натуральный логарифм мольных долей паров; - натуральный логарифм мольных долей паров;
NC – число компонентов;
NT – число тарелок.
Подстрочные:
i – индекс тарелки;
j – индекс комнонента.
Надстрочные:
F – относится к сырью;
D – относится к выводу продукта;
L – относится к свойствам жидкости;
V – относится к свойствам паров.
Неизвестные, иначе называемые итерационными или первичными переменными: (X, Y, L, V, T,)I, где I = 1, NT, определяются при непосредственном использовании метода приращений Ньютона – Рафсона. Уравнения спецификаций, включающие в себя итерационные переменные, добавляются непосредственно к приведенным выше уравнениям и решаются совместно.
Имеется две модификации метода Ньютона – Рафсона. Первая представляет собой процедуру линейного поиска, которая будет, где это возможно, уменьшать сумму ошибок коррекции Ньютона. Если это не представляется возможным, размер увеличения будет органичен предварительно заданным значением. Вторая модификация ограничивает изменения отдельных итерационных переменных. Обе эти модификации могут в результате привести к дробному движению в направлении Ньютоновского решения. Дробный размер шага выводится в суммарном отчете по итерациям колонны.
4.2. Синтез технологических схем экстрактивной ректификации в комплексах простых и сложных колонн.
Азеотропным смесям присущи термодинамико-топологические ограничения на выделение конечных фракций заданного состава. Основой для преодоления ограничений служит принцип перераспределения полей концентраций между областями разделения [23–25]. Этот принцип может быть реализован с использованием однородных и неоднородных (с включением экстракции, адсорбции, абсорбции, химических и др. методов разделения) разделительных комплексов. В первом случае процесс ректификации может быть организован так, что на одном из этапов осуществляется выделение азеотропной фракции, которая затем подвергается разделению с использованием специальных методов. Если это фракция двух и более компонентов, то можно использовать экстрактивную ректификацию или разделение азеотропного состава под разными давлениями. Если фракция содержит более двух компонентов, появляется возможность применить для разделения методы, использующие кривизну разделяющего многообразия [25, 26]. В работах [24, 27] предложен общий подход к синтезу схем ректификации многокомпонентных неидеальных (включая азеотропные) смесей, основанный на понятии области ректификации. Авторами [28–30] предложены некоторые подходы к синтезу технологических схем ректификации многокомпонентных смесей, содержащих один бинарный азеотроп. При этом используется метод разделения азеотропной смеси под разными давлениями.
Наиболее часто на практике для преодоления термодинамико-топологических ограничений на выделение конечных фракций заданного состава используют метод экстрактивной ректификации. Он играет важную роль при разделении сложных азеотропных смесей. Традиционные схемы экстрактивной ректификации обладают значительной энергоемкостью. Поэтому разработка и применение оптимальных энергосберегающих технологий является актуальной задачей. Ее решение включает в себя несколько этапов:
1. Структурная оптимизация технологической схемы.
2. Оптимизация рабочих параметров ректификационных колонн.
3. Конструкционная оптимизация элементов технологической схемы.
Поскольку процесс ректификации является необратимым, то его термодинамическая эффективность зависит от пути. В качестве таковых можно рассматривать наборы технологических схем или траектории ректификации. Подходы к синтезу технологических схем экстрактивной ректификации в настоящее время разработаны явно недостаточно. Этому вопросу посвящены только отдельные работы. Например, в [31] предложен ряд комплексов с различной структурой, обеспечивающих разделение многокомпонентных азеотропных смесей. Авторами [32] предложен ряд технологических решений по ректификации смеси этанол – вода. Однако в ряде случаев в работе не учтены термодинамико–топологические ограничения на составы продуктов и предлагаемые решения неработоспособны. Таким образом, в настоящее время практически за пределами рассмотрения остается вопрос структурной оптимизации процесса экстрактивной ректификации. Решение этой проблемы целесообразно начать с разработки общих алгоритмов синтеза технологических схем.
К настоящему времени сложилась классификация схем экстрактивной ректификации. Все возможные варианты разделения можно разбить на две большие группы [31]. К первой группе относятся схемы, в которых уже на первом этапе разделения применяется экстрактивный агент и, соответственно, снимаются термодинамико–топологические ограничения на составы продуктовых фракций. Вторая группа характеризуется предварительным фракционированием исходной многокомпонентной смеси вплоть до выделения фракции азеотропообразующих компонентов. Затем эту фракцию разделяют экстрактивной ректификацией.
В целом, эти два класса схем охватывают все возможные варианты разделения. Схемы из первого класса для ректификации смесей с низкой размерностью концентрационного пространства можно использовать как элементы разделения смесей во второй группе схем для смесей более высокой размерности.
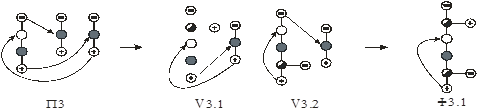
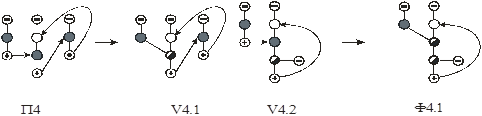
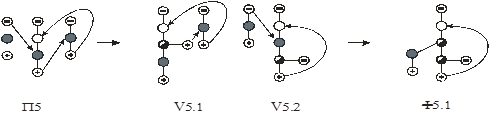
На первом этапе рассмотрен алгоритм синтеза схем первого класса. Поскольку введение экстрактивного агента снимает термодинамико–топологические ограничения на составы продуктовых потоков, то в качестве прообразов для синтеза использованы наборы схем ректификации зеотропных смесей из простых двухсекционных колонн. Принято, что разделению подвергается (n+1)–компонентная смесь, состоящая из n–компонентной исходной азеотропной смеси и экстрактивного агента. Предположено, что экстрактивный агент является самым тяжелолетучим компонентом в смеси.
На рис. 10 представлены схемы ректификации для трех– и четырехкомпонентных зеотропных смесей.

Рис. 10. Схемы разделения (а, б) – трех- и (в-ж) – четырехкомпонентной зеотропной смеси. А, В, С – компоненты смеси,
S
– тяжелокипящий агент,
F
– питание АВ
S
для схем (а, б) и АВС
S
– для остальных.
Для последующего анализа схемы представлены в виде ориентированных графов с вершинами – колоннами и фракциями, ребрами – потоковыми связями между ними (рис. 10). Вершины – фракции помечены соответствующими буквами. Так, например, схема рис. 10в будет иметь вид, представленный на рис. 11.
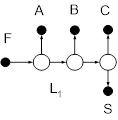 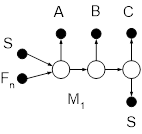
Рис.11. Представление схемы ректификации в виде орграфа
L
.  – вершины–колонны; – вершины–колонны;  – вершины-фракции – вершины-фракции
Рис. 12. Граф М, полученный расщеплением вершины у графа
L
Представленная на рис. 11 структура отображает последовательность разделения. Следующим этапом в синтезе является расщепление потока F (n+1)-компонентной смеси на два индивидуальных потока – n-компонентную смесь Fn и экстрактивный агент S. Формально, если базироваться на структуре графа L рис.11, получается граф М на рис. 12.
Данная структура уже почти в полной мере отображает схему экстрактивной ректификации. Требуется только применить цикл по экстрактивному агенту S, отождествив (стянув в одну) две вершины – фракции с индексом S. Получен граф U рис. 13.
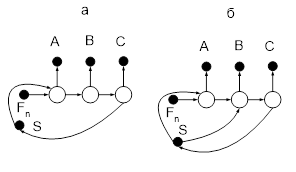
Рис. 13. Граф
U
, полученный отождествлением одноименных вершин – фракций графа М.
В зависимости от структуры паро-жидкостного равновесия из вершины S может исходить не единственное ребро (экстрактивный агент может понадобиться и для разделения смеси во второй колонне комплекса) (рис. 13б).
При этом мы допускается, что имеется такой экстрактивный агент S, который позволит обеспечить разделение всей n-компонентной смеси на чистые компоненты.
Поскольку комплекс экстрактивной ректификации имеет замкнутый (без учета потерь) цикл по экстрактивному агенту, то одним из условий работоспособности синтезированных по предложенному алгоритму схем будет наличие в орграфе цикла. Под этим термином подразумевается возможность обхода ряда вершин графа с возвращением в исходную вершину с учетом направленности ребер. При этом цикл должен иметь в своем составе, по крайней мере, две вершины, эксплицирующих колонны. Вторым условием работоспособности предложенных схем является ввод экстрактивного агента в колонну разделения азеотропной пары компонентов.
Далее представлен наиболее простой пример – разделение бинарной азеотропной смеси АВ.
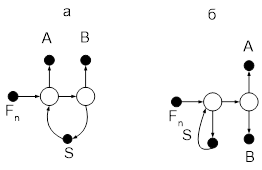
Рис. 14 Графы схем экстрактивной ректификации азеотропной смеси АВ
Для рассматриваемого типа схем экстрактивный агент должен подаваться в первую колонну разделения, поэтому на рис. 14 приведены только эти варианты.
Видно, что условию работоспособности отвечает только граф на рис. 14а. Он полностью структурно соответствует классической схеме экстрактивной ректификации бинарной азеотропной смеси с тяжелолетучим разделяющим агентом.
Общий подход к синтезу технологических схем разделения для четырехкомпонентной азеотропной смеси можно представить в следующем виде:
1. Синтез схемы ректификации четырехкомпонентной зеотропной смеси (рис. 10 в-ж) и представление их в форме орграфов типа L(рис. 15 а-д).
2. Выделение в существующем наборе схем (рис. 15 а-д) вершин, в которые необходимо направить ребро из S (экстрактивная колонна в схеме). Для выбранного типа схем экстрактивной ректификации это вершины – колонны, первые по ходу разделения. Выделяются темным кружком.
3. Расщепление вершины F на Fn
и S, удаление вершины S, инциндентную исходящему ребру. Эта операция эквивалентна замене вершины F на Fn
.
4. Связывание вершины S с помеченной вершиной-колонной ориентированным исходящим из S ребром и проверка полученной структуры (рис. 15 л-о) на работоспособность исходя из портрета паро-жидкостного равновесия в соответствии с классификацией Л.А. Серафимова [33].
5.
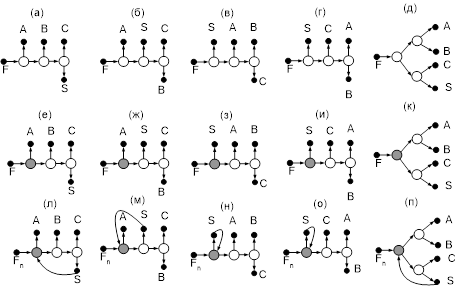
Рис. 15. Синтез графов – схем экстрактивной ректификации
Доказано, что применение сложных колонн при экстрактивной ректификации (ЭР) [35, 36] приводит к повышению термодинамической эффективности процесса и снижению энергозатрат на разделение [37–39]. Решение задачи синтеза схем ректификации зеотропных смесей с частично связанными тепловыми и материальными потоками предложено в работе [40]. Для определения работоспособных вариантов ЭР использовался алгоритм [40], дополненный его запретом на стягивание по ориентированному ребру. Для реализации алгоритма [40] потребовалось представить технологические схемы в виде графов. Однако в этом случае вершины соответствуют сечениям, разделяющим либо ограничивающим секции колонн, а ребра — потокам пара и жидкости. Пара разнонаправленных ребер, инцидентных одной паре вершин, отображает секцию колонны (рис. 16).
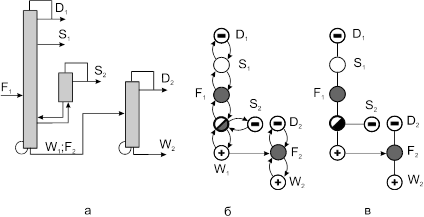
Рис. 16. Представление технологической схемы ректификации в иконографическом виде – (а), в виде ориентированного мультиграфа – (б), в виде ориентированного графа – (в);  — питание (вход), — питание (вход),  — отбор (выход) с отводом тепла, — отбор (выход) с отводом тепла,  — отбор (выход) с подводом тепла, — отбор (выход) с подводом тепла,  — отбор (выход) без подвода или отвода тепла, — отбор (выход) без подвода или отвода тепла,  — вершина, не обладающая свойством входа или выхода. — вершина, не обладающая свойством входа или выхода.
Далее компоненты смеси (обозначенные А, В, С) расположены в порядке увеличения температур кипения. Экстрактивный агент — самый тяжелокипящий компонент смеси (S). В работе [34] выявлены работоспособные «базовые» варианты организации процесса ЭР трехкомпонентных азеотропных смесей. Всего таких вариантов пять. Три организованы таким образом, что экстрактивный агент направляется в первую колонну и, при необходимости, — во вторую.
Вариант П1 (рис.17 а)
состоит из трех двухотборных колонн, целевым продуктом в каждой из которых является легколетучий компонент. Первая колонна — колонна экстрактивной ректификации. Подача ЭА осуществляется из куба третьей колонны в верхнее сечение первой колонны. В схемах П2 и П3 (рис.17 б
,в
) колонна ЭР также расположена первой по ходу разделения. В первой колонне схемы П2 осуществляется выделение под действием ЭА самого легколетучего компонента А, во второй колонне происходит выделение экстрактивного агента и, наконец, в последней колонне — разделение средне- и тяжелокипящих компонентов. В схеме П3 за колонной экстрактивной ректификации также следуют две колонны, однако под действием ЭА осуществляется разделение смеси на две фракции АВ и CS, каждая из которых затем разделяется в индивидуальных колоннах.
Рис.16 Преобразование графов традиционных схем экстрактивной ректификации в графы схем с частично связанными тепловыми и материальными потоками.
Существуют еще две схемы из двухотборных колонн П4 и П5 (рис.17. г,д
). Разделение в них основано на предварительном отделении зеотропной составляющей трехкомпонентной смеси без применения экстрактивного агента. В схеме П4 — это предварительное выделение легкокипящего компонента А, а в схеме П5 — тяжелокипящего компонента С. Затем бинарная смесь ВС в схеме П4 и АВ в схеме П5 разделяется под действием разделяющего агента S в традиционном комплексе ЭР.
Применение алгоритма [40] к схемам прообразам П1—П3 позволяет получить по три схемы образа для каждой из них. Образом схемы П1 являются схема V1.1, состоящая из сложной колоны экстрактивной ректификации. Вплоть до зоны питания эта структура полностью соответствует классической схеме ЭР с тяжелолетучим агентом. Ниже зоны питания расположена укрепляющая боковая секция. Тяжелокипящие продукты направляются в простую двухсекционную колонну регенерации экстрактивного агента. Эта колонна полностью идентична третьей колонне схемы П1. Стягивание графа, эксплицирующего схему П1, по ориентированному ребру, связывающему подграфы, отображающие вторую и третью колонны схемы, приводит к варианту организации процесса по графу V1.2. В этом случае экстрактивная колонна не подвергается трансформации. Преобразуются только вторая и третья по ходу разделения колонны, образующие комплекс с частично связанными тепловыми и материальными потоками в виде колонны с боковой укрепляющей секцией, расположенной ниже зоны питания. Более глубокие преобразования приводят к одной сложной колонне, в которой рецикл экстрактивного агента связывает куб и верхнюю часть колонны, а ниже зоны питания расположены две укрепляющие секции. В реальной схеме должен присутствовать теплообменник, обеспечивающий требуемую температуру подачи экстрактивного агента в колонну ЭР. Однако поскольку его расположение топологически неизменно по отношению к колонне ЭР, то при синтезе схем на данном этапе исследования он не учитывался.
Схема П3 является прообразом для двух схем класса V (V3.1, V3.2). В схеме V3.1 в первой колонне экстрактивной ректификации выше зоны ввода экстрактивного агента расположена укрепляющая боковая секция для выделения компонента В. Колонна регенерации ЭА остается без изменений относительно П3. В схеме V3.2 в качестве дистиллята колонны экстрактивной ректификации отбирают смесь компонентов А и В, разделяемых в простой колонне. Схема-образ V3.3 состоит из одной сложной колонны, охваченной рециклом экстрактивного агента от куба до экстрактивной секции. Выше экстрактивной зоны расположена боковая исчерпывающая секция, ниже зоны питания — боковая укрепляющая секция.
Образы схемы П2 более сложны. В схеме V2.1 в первой колонне протекает как процесс выделения компонента А экстрактивной ректификацией, так и регенерация экстрактивного агента, выделяемого в качестве кубового продукта. Боковой погон, в основном состоящий из компонентов В и С и выделяемый из укрепляющей секции, которая расположена ниже зоны питания, направляется на ректификацию в простую колонну.
Отмечено, что схема П1 с единственным рециклом экстрактивного агента, обеспечивает за счет экстрактивной ректификации только выделение самого легкокипящего компонента А. Именно такой режим поддерживают и схемы-образы V1.1, V1.2, F1.1. В случае, если требуется применение экстрактивного агента для разделения компонентов В и С, то необходимо и во второй колонне проводить экстрактивную ректификацию. Это потребует организации второго рециркуляционного потока по экстрактивному агенту. Обозначив схемы с двумя рециклами по экстрактивному агенту дополнительным индексом d, на основе V1.1 получены V1.1d и т.д.(рис.18). Организация процесса экстрактивной ректификации по вариантам V1.1d и F1.1d требует отдельной экспериментальной и расчетной проработки, поскольку в литературе до настоящего времени отсутствуют данные о применимости двухуровневой подачи экстрактивного агента. Работоспособность схемы на основе графа V1.2d не вызывает сомнений, поскольку уже к настоящему времени показана работоспособность и эффективность таких схем ректификации [35].
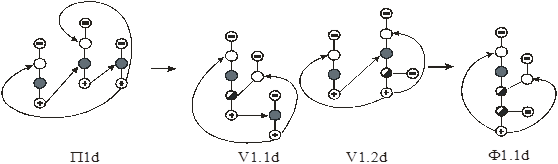
Рис.18 Граф схемы экстрактивной ректификации с подачей экстрактивного агента в две колонны комплекса П1 и его образы, включающие сложные колонны.
В соответствии с исходной структурой схемы-образы на основе П2 казалось бы не пригодны для организации двух рециркуляционных потоков по экстрактивному агенту. Это связано с тем, что требуется охват этим потоком всего комплекса. Однако, поскольку операции стягивания по ориентированному ребру могут затрагивать все ориентированные ребра за исключением, эксплицирующих рециклы, то в схемах V2.2d и F2.1d требование охвата одним из рециклов всего комплекса выполняется также, как и для V2.2 и F2.1. То же относится и к образам схемы П3. Работоспособными могут считаться схемы V3.1d и F3.5d при допущении о применимости двухуровневой подачи экстрактивного агента.
Схемы с предварительным фракционированием П4 и П5 (рис.2 в,г
) отличаются типом разделения в первой по ходу колонне. В П4 — это выделение самого легколетучего компонента А простой ректификацией, в П5 — самого тяжелокипящего компонента С. Оставшуюся пару компонентов разделяют в обычном двухколонном комплексе ЭР.
Процедура графовой трансформации, (как и в [37]) приводит к получению трех структур для каждого из прообразов, т.е. V4.1, V4.2, F4.1 для П4 и V5.1, V5.2, F5.1 для П5. Для всех этих схем характерен охват рециркуляционным потоком экстрактивного агента только части колонн комплекса. Эта особенность сохраняется и при операциях трансформации, следовательно, все эти типы схем не могут реализовать в своем составе два рецикла по экстрактивному агенту. На основе трансформации схемы П4 (рис.2 в
) получаем схему V4.1, в которой исходная смесь подается в двухсекционную колонну (предфракционатор), имеющую конденсатор и связанную тепловыми и материальными потоками по нижнему сечению с колонной экстрактивной ректификации. Кубовый продукт этого комплекса направляется в колонну регенерации экстрактивного агента. В схеме V4.2 первой по ходу разделения расположена двухсекционная колонна выделения компонента А. Смесь компонентов В и С разделяют в сложной колонне экстрактивной ректификации с боковой укрепляющей секцией, расположенной ниже зоны исходного питания. Схема F4.1 состоит из одной сложной колонны, имеющей двухсекционный префракционатор, служащий для выделения компонента А и соединенный в своем нижнем сечении потоками пара и жидкости с основной колонной ниже зоны подачи экстрактивного агента. Между зоной стыковки префракционатора и кубом основной колонны присоединена боковая укрепляющая секция для выделения компонента С. Дистиллят основной колонны — компонент В. Кубовый продукт — экстрактивный агент S — рециркулируют в верхнюю часть основной колонны. Все варианты работоспособны. Структура F4.1 позволяет также организовать процесс разделения экстрактивной ректификацией и пары компонентов АВ, поскольку имеется возможность реализовать в предфракционаторе выше зоны питания экстрактивную секцию.
При трансформации схемы П5 (рис.17 г
) также получены три типа новых структур V5.1, V5.2, F5.1. Поскольку в результате процедуры стягивания в схеме П5 по первому по ходу разделения ориентированному ребру образуются комплексы с частично связанными тепловыми и материальными потоками, имеющие по два кипятильника и один дефлегматор, то разделение компонента С и экстрактивного агента S с заданной чистотой становится проблематичным. Такая трансформация лежит в основе синтеза схем V5.1 и F5.1, следовательно они не работоспособны. Таким образом, единственной работоспособной схемой-образом П5 с частично связанными тепловыми и материальными потоками является V5.2. В ней на первом этапе в простой двухсекционной колонне отделяется обычной ректификацией компонент С, а разделение пары АВ производится экстрактивной ректификацией в сложной колонне с боковой укрепляющей секцией, которая служит для выделения компонента В и расположена ниже зоны питания. [34]
5.
Расчетно - экспериментальная часть
Данные о физико – химических свойствах бензола, циклогексана, гексан и сульфолан взяты из базы данных программы PRO/II фирмы SIMSCI.
Таблица 4.4.
| Наименование вещества |
Молекулярная масса;
г/моль
|
Относительная плотность; кг/м3
|
tкип
;
0
С
|
| бензол |
78,1150 |
883,92 |
80,1 |
| гексан |
| циклогексан |
84,163 |
782,65 |
80,74 |
| сульфолан |
На первом этапе исследований перед нами стоит задача выбора модели, адекватно описывающей ПЖР в четырехкомпонентной смеси бензол – толуол – циклогексан – ДМФА. Для проведения оценки адекватности нами были собраны имеющиеся в литературе данные о фазовом равновесии для бинарных составляющих смеси [44-46]:
бензол – ЦГ
гексан - ЦГ
бензол – гексан
бензол – ДМФА
(для систем толуол – ДМФА и циклогексан – ДМФА экспериментальных данных нет).
Также для всех 6 групп бинарных систем были получены псевдоэкспериментальные данные при помощи групповой модели UNIFAC.
На основании общих рекомедаций были выбраны две группы математических моделей, основанных на различных представлениях о структуре раствора и межмолекулярных взаимодействий:
· Модели локальных составов (Wilson, NRTL, UNIQUAC)
· Уравнения состояния (SRK, Peng - Robinson)
Для статистической обработки результатов в качестве критерия, оценивающего адекватность описания ПЖР, выбрана средняя относительная погрешность описания экспериментальных данных различными моделями δ:
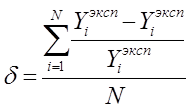 , N – количество точек фазового равновесия. 5.1. , N – количество точек фазового равновесия. 5.1.
Результаты статистической обработки имеющихся псевдоэкспериментальных данных представлены в таблицах 2 и 3:
Таблица 2
Средняя относительная погрешность описания экспериментальных данных различными моделями
| бинарная пара |
δ |
| NRTL |
UNIQUAC |
Wilson |
SRK |
PR |
UNIFAC |
| бензол - ЦГ |
0,0300 |
0,0283 |
0,0237 |
0,0482 |
0,0644 |
0,0315 |
| ЦГ - толуол |
0,0499 |
0,0489 |
0,0516 |
0,1907 |
0,1333 |
0,0220 |
| бензол - толуол |
0,1041 |
0,0689 |
0,0977 |
0.0467 |
0,0658 |
0,0160 |
| бензол - ДМФА |
0,2443 |
0,1987 |
0,2147 |
0,2363 |
0,2180 |
0,0500 |
Таблица 3. Средняя относительная погрешность описания псевдоэкспериментальных данных, полученных при помощи групповой модели UNIFAC, различными моделями
| бинарная пара |
δ |
| NRTL |
UNIQUAC |
Wilson |
SRK |
PR |
| бензол - ЦГ |
0,1019 |
1,11∙10-5
|
5,32∙10-3
|
0,0343 |
0,0714 |
| бензол - толуол |
8,79∙10-3
|
5,24∙10-5
|
4,48∙10-3
|
0,0152 |
0,0576 |
| бензол - ДМФА |
0,011 |
9,34∙10-5
|
0,0551 |
0,1237 |
0,1041 |
| толуол - ДМФА |
0,0741 |
0,0634 |
0,0803 |
0,0486 |
0,0672 |
| толуол - ЦГ |
0,0496 |
0,0561 |
0,0572 |
0,0441 |
0,0876 |
| ЦГ – ДМФА* |
- |
- |
- |
- |
- |
*- из-за высокой неидеальности системы ЦГ-ДМФА оценить адекватность модели при помощи δ невозможно.
Как можно заметить из приведенных выше результатов все выбранные модели удовлетворяют условиям адекватности, однако нам необходима такая модель, которая будет способна описать сложный фазовый портрет в системе ЦГ-ДМФА. Поэтому казалось бы целесообразным применить для расчета групповую модель UNIFAC, которая удовлетворительно описывает экспериментальные данные и хорошо согласуется с данными, полученными по другим моделям (табл. 3).
Однако, поскольку в системе может реализоваться сосуществование двух жидких фаз, то этот вопрос также требует обсуждения и оценки применимости той или иной модели.
Исходя из литературных данных, система характеризуется сильным отклонением от идеальности обусловленным наличием азеотропа и расслаивания. Согласно информации, полученной нами из банка данных по физико-химическим свойствам компонентов и их смесей «RSA DB» Решетова С. А. при атмосферном давлении вторая жидкая фаза присутствует в интервале температур от 20 до 53 ºС.
5.3. Критерий оптимизации.
В качестве критерия при подборе оптимальных рабочих параметров колонн мы использовали минимум суммарных энергетических затрат в кипятильниках колонн:
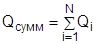
где: N- число колонн в технологической схеме.
Энергетические затраты в кубе каждой колонны рассчитываются исходя из общего теплового баланса.
Для колонны экстрактивной ректификации уравнение теплового баланса имеет вид:
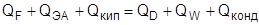 (5.2) (5.2)
где:
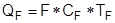 - количество тепла, поступающее с потоком исходной смеси; - количество тепла, поступающее с потоком исходной смеси;
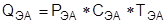 - количество тепла, поступающее в колонну с потоком экстрактивного агента; - количество тепла, поступающее в колонну с потоком экстрактивного агента;
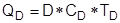 - количество тепла, отводимое из колонны с потоком дистиллята; - количество тепла, отводимое из колонны с потоком дистиллята;
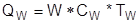 - количество тепла, отводимое из колонны с кубовым потоком; - количество тепла, отводимое из колонны с кубовым потоком;
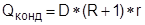 - количество тепла, отводимое при конденсации потоков дистиллята и флегмы; - количество тепла, отводимое при конденсации потоков дистиллята и флегмы;
Откуда затраты тепла в кипятильнике:
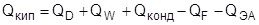 (5.3) (5.3)
или
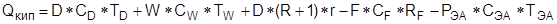 (5.4) (5.4)
При заданном количестве и составе питания, а также заданном качестве продукта, поток дистиллята определяется из общего материального баланса колонны и является фиксированной величиной. Следовательно, теплосодержание верхнего продукта QD
есть величина практически постоянная. Величина кубового потока также определяется из материального баланса и зависит от соотношения исходная смесь : ЭА. Следовательно, теплосодержание нижнего продукта QW
зависит от расхода разделяющего агента.
При проведении расчетов мы полагали, что исходная смесь поступает в колонну при температуре кипения.
Таким образом, величина энергетических затрат в кубе экстрактивной колонны в данном случае зависит в основном от флегмового числа, температуры и расхода подаваемого в колонну экстрактивного агента.
Как известно, флегмовое число в колонне заданной эффективности определяется, с одной стороны, природой разделяемой смеси, а с другой, положением тарелки питания, поскольку в зависимости от точки подачи исходной смеси в колонне формируется определенный профиль концентраций компонентов (траектория ректификации).
В колонне экстрактивной ректификации на формирование профиля оказывает существенное влияние и уровень ввода экстрактивного агента.
Температура подачи в колонну экстрактивного агента оказывает неоднозначное влияние на величину энергозатрат. С одной стороны, как видно из уравнения (4.3), чем выше температура экстрактивного агента, тем меньше на первый взгляд Qкип
. Однако, поскольку экстрактивный агент подают в верхнее сечение колонны при этом увеличивается его концентрация на верхних тарелках, и для получения дистиллята заданного качества потребуются более высокие значения флегмового числа, что, в свою очередь, приводит к росту Qкип
.
Для колонны регенерации ацетонитрила уравнение теплового баланса имеет вид:
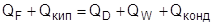
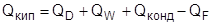 (5.5) (5.5)
(обозначения те же, что и в формуле 5.2)
Количество питания, потоки дистиллята и куба в этой колонне при фиксированном соотношении исходная смесь : экстрактивный агент и заданном качестве продуктовых потоков определяются из общего материального баланса схемы, и являются постоянными величинами. Поэтому в данном случае Qкип
зависит в основном от флегмового числа, которое, в свою очередь, определяется положением тарелки подачи в колонну исходной смеси.
Таким образом, существует определенный набор рабочих параметров, при котором энергозатраты на разделение минимальны.
· Для колонны экстрактивной ректификации такими параметрами являются: взаиморасположение тарелок подачи в колонну исходной смеси и экстрактивного агента, температура и расход экстрактивного агента, флегмовое число.
· Для колонны регенерации экстрактивного агента: уровень ввода исходной смеси и флегмовое число.
 5.4 Анализ работоспособности синтезированных схем разделеня.
5.4 Анализ работоспособности синтезированных схем разделеня.

Рис.18 Портрет паро-жидкостного равновесия смеси ЦГ-бензол-толуол
Система ЦГ-бензол-толуол характеризуется наличием азеотропа с минимум Ткип на стороне бензол-ЦГ, сепаратрисса azбензол-ЦГ
– толуол делит концентрационный симплекс на две области дистилляции ЦГ- azбензол-ЦГ
– толуол и azбензол-ЦГ
– бензол – толуол.
Работоспособные схемы:
П1 (V1.1, V1.2, Ф1.1), П2 (V2.1, V2.2, Ф2.1), (см. 3.2.)– на первом этапе экстрактивной ректификацией выделяется компонент бензол, далее разделяется зеотропная составляющая ЦГ-толуол.
П5 (V5.2) – на первом этапе предварительным фракционированием выделяется компонент толуол, далее экстрактивной ректификацией разделяется азеотропная пара бензол-ЦГ.
Неработоспособные схемы
:
П3, V3.2 – в первой колонне в качестве дистиллята выделяется азеотропная составляющая бензол-ЦГ, которую невозможно разделить простой ректификацией.
V3.1, Ф3.1 – невозможно разделить азеотропную составляющую бензол-ЦГ, так как точки отбора этих компонентов находятся выше зоны экстрактивной ректификации (точки подачи ЭА), организация двухуровневой подачи экстракивного агента не решит задачу, так как приведет к попаданию ЭА в боковую исчерпывающую секцию и не позволит выделить чистый компонент ЦГ.
П4 (V4.1, V4.2, Ф4.1) – на первом этапе выделить предварительным фракционироанием компонент бензол невозможно, так как он образует азеотроп с компонентом ЦГ.
5.5 Расчет схем экстрактивной ректификации.
На первом этапе мы рассчитывали энергозатраты на разделение в простых колоннах. Согласно проведенному нами выше анализу существует три работоспособных варианта разделения исходной смеси в схемах состоящих из простых двухсекционных колонн. (рис.19)
Рис.19. Схемы, состоящие из простых двухсекционных колонн (П1, П2, П5)
 а) Схема П1 а) Схема П1
б) Схема П2

Все расчеты проводили на 1000 кг/час исходной смеси, содержащей 37% масс. ЦГ, 23% масс. бензола, 40% масс. толуола. Полагали также что поток РА содержит 99,96 масс. ДМФА и 0,04 масс. толуола, попадающего в поток РА вместе с рециклом из колонны регенерации.
Параметры работы колонн, расхода РА и число тарелок подбирались методом последовательного сканирования так, чтобы обеспечить наилучшее качество продуктовых потоков. Процесс проводится при условиях вакуума (P=0,1 атм) для того чтобы сдвинуть границу тангециального азеотропа ДМФА – циклогексан (95% масс. ДМФА) и для предотвращения термического разложения РА. [43]
Энергозатраты на разделение и параметры процесса представлены в табл.5, а материальный баланс в Приложении 2.
Таблица 5.
Рабочие параметры простых схем экстрактивной ректификации.
| П1 |
П2 |
П5 |
| колонна |
Т1 |
Т2 |
Т3 |
Т1 |
Т2 |
Т3 |
Т1 |
Т2 |
Т3 |
| Qконд,
Гкал/час. |
-0,3350 |
-0,2053 |
-0,4191 |
-0,3355 |
-0,2786 |
-0,0643 |
-0,1137 |
-0,3653 |
-0,0747 |
| Qкип,
Гкал/час |
0,2791 |
0,4386 |
0,2306 |
0,2786 |
0,3144 |
0,0634 |
0,0876 |
0,5440 |
0,1036 |
| R |
8,5 |
8,6 |
9,9 |
5 |
3,3 |
1,7 |
0,3 |
15,22 |
2,1623 |
| NЭА
/NF
|
11/14 |
14 |
15 |
11/14 |
9 |
11 |
10 |
7/12 |
1 |
| Nт.т.
|
20 |
20 |
20 |
20 |
20 |
20 |
20 |
20 |
20 |
| Q∑
кип,
Гкал/час |
0,9483 |
0,6564 |
0,7352 |
На втором этапе мы производили расчет схем ректификации, состоящих из последовательности простая двухсекционная колонна – колонна с боковой секцией и сложная колонна с двумя боковыми секциями. Расчетный эксперимент показал, что в колоннах наблюдается расслаивание жидкой фазы, а значит затруднительно обеспечить равномерный отбор брутто-составов жидкости с тарелки. Поэтому варианты V2.2, Ф2.1, подразумевающие отбор в боковую секцию жидкой фазы, были исключены из расчета. Рассчитанные схемы с частично связанными тепловыми и материальными потоками представлены на рис. 20.
Рис.20. Схемы с частично связанными тепловыми и материальными потоками (V1.1, V1.2, Ф1.1, V2.1, V5.2)
 а1
) схема V1.1 а2
) схема V1.2 а3
) схема Ф1.1 а1
) схема V1.1 а2
) схема V1.2 а3
) схема Ф1.1
б) схема V2.1

в) схема V5.2
При расчете синтезированных сложных схем, мы закрепляли число тарелок в секциях, а также положение тарелок питания и подачи РА. Параметры работы схем V1.1, V1.2, Ф1.1, V2.1, V5.2 представлены в табл.6-8.
Taблица 6. Рабочие параметры схем-образов П1*
| V1.1 |
V1.2 |
Ф1.1 |
| колонна |
Т1 |
S1.1 |
Т2 |
Т1 |
Т2 |
S2.1 |
Т1 |
S1.1 |
S1.2 |
| Qконд,
Гкал/час. |
-0,274 |
-0,1866 |
-0,4191 |
-0,3355 |
-0,2350 |
-0,1544 |
-0,2113 |
-0,1388 |
-0,1900 |
| Qкип,
Гкал/час |
0,5943 |
- |
0,2306 |
0,2791 |
0,6269 |
- |
0,5287 |
- |
- |
| R |
5,2 |
8,6 |
9,9 |
5 |
10 |
2,9 |
3,1 |
3,4 |
3,9 |
| NЭА
/NF
|
11/14 |
-/14 |
-/15 |
11/14 |
-/14 |
-/15 |
11/14 |
-/14 |
-/15 |
| NS1
/NS2
|
20/- |
- |
- |
- |
20/- |
- |
20/26 |
- |
- |
| Nт.т.
|
26 |
14 |
20 |
20 |
25 |
15 |
31 |
14 |
15 |
| Q∑
кип,
Гкал/час |
0,8249 |
0,906 |
0,5287 |
*NS1
- тарелка отбора в первую боковую секцию; S1 – первая боковая секция; NS2
- тарелка отбора в первую боковую секцию; S2 –вторая боковая секция
∆ - снижение энергозатрат схем образов относительно схем праобразов, %
Taблица 7. Рабочие параметры схемы-образа П2 (
V
2.1)*
| колонна |
Т1 |
S1.1 |
Т2 |
| Qконд,
Гкал/час. |
-0,1933 |
-0,2311 |
-0,0643 |
| Qкип,
Гкал/час |
0,4043 |
- |
0,0634 |
| R |
4,7 |
2,6 |
1,7 |
| NЭА
/NF
|
11/14 |
-/9 |
-/11 |
| NS1
|
20 |
- |
- |
| Nт.т.
|
31 |
9 |
20 |
| Q∑
кип,
Гкал/час |
0,4677 |
Taблица 8. Рабочие параметры схемы-образа П5 (V5.2)*
| колонна |
Т1 |
Т2 |
S2.1 |
| Qконд,
Гкал/час. |
-0,1137 |
-0,1649 |
-0,0304 |
| Qкип,
Гкал/час |
0,0876 |
0,2200 |
- |
| R |
0,9 |
5,8 |
0,7 |
| NЭА
/NF
|
-/10 |
7/12 |
-/10 |
| NS1
|
- |
20 |
- |
| Nт.т.
|
20 |
30 |
10 |
| Q∑
кип,
Гкал/час |
0,3076 |
5.6. Обсуждение полученных результатов
Для сравнительного анализа полученных результатов в табл.9 представлены суммарные энергозатраты для всех рассмотренных схем.
Таблица 9.
Сводная таблица полученных результатов по всем рассмотренным схемам.
| П1
|
V1.
1
|
V1.
2
|
Ф1.1
|
П2
|
V
2.1
|
П5
|
V
5.2
|
| Q∑
кип,
Гкал/час
|
0,9483 |
0,8249 |
0,9060 |
0,5287 |
0,6564 |
0,4677 |
0,7352 |
0,3076 |
| ∆, %
|
-
|
13,01 |
4,06 |
44,25 |
-
|
28,75 |
-
|
58,16 |
Как видно из табл.9 применение связывания тепловых и материальных потоков приводит к существенному снижению суммарных энергозатрат на разделение в процессе экстрактивной ректификации.
Среди образов схемы П1 наименьшими энергозатратами обладает образ Ф1.1, он на 0,4196 Гкал/час (44,25%) менее энергоемок схемы праобраза, на 0,2962 Гкал/час (35,91%) менее энергоемок схемы образа V1.1 и на 0,3773 Гкал/час (41,64%) менее энергоемок схемы образа V1.2. Образ схемы П2V2.1 на 0,1887 Гкал/час (28,75%) менее энергоемок схемы праобраза. И наконец образ схемы П5 V5.2 на 0,4276 Гкал/час (58,16%) менее энергоемок схемы праобраза.
Казалось бы, что среди всех рассмотренных схем, образ Ф1.1. наиболее приближен к термодинамически обратимому процессу, и следовательно он должен обладать минимальными энергозатратами. Однако расчет показал, что наименьшими тепловыми нагрузками обладает схема V5.2. (Q∑кип
= 0,3076 Гкал/час). Это можно объяснить тем, что рассмотренная нами смесь содержит значительно количество тяжелокипящего толуола, поэтому наиболее эффективной оказалась схема с предварительным отделением тяжелокипящего компонента от бинарной азеотропной составляющей.
6. Выводы.
· Для рассматриваемой системы уменьшение числа аппаратов, охваченных рециклом, приводит к значительному уменьшению суммарных энергозатрат в кипятильниках колонн на 4 - 58%. Это связано с меньшими затратами тепла в кубах колонн в соответствии с уменьшением количества разделяемого агента.
· В ряду схем образов первого заданного разделения наилучшими показателями обладает схема, имеющая минимальное число кипятильников (Ф 1.1). При этом экономия энергоресурсов по сравнению с праобразом П1 составляет 44,25%.
· Из трех возможных схем, состоящих из двух отборных колонн, схемы работающие по второму заданному разделению и разветвленная система обладают относительно небольшим отличием в энергозатратах (10,72%)
· В связи с наличием двух жидких фаз при ректификации в качестве пригодных для промышленной реализации могут быть использованы только по одному образу П2 и П3. Разница в энергозатратах между образами этих схем составляет 41,28% по отношению к друг другу и наименьшими энергозатратами характеризуется схема V 3.2, имеющая энергозатраты на 66% меньше, лучшего варианта из отборных колонн П2.
1. Тимофеев В.С., Серафимов Л.А «Принципы технологии основного органического и нефтехимического синтеза», Москва «Высшая школа» 2003 г, 536 с
2. Сулимов А. Д. «Производство ароматических углеводородов из нефтяного сырья», Москва, 1975 г.
3. Zhigang Lei, Chengyue Li, & Biaohua Chen. Extractive distillation: a review. Separation and purification reviews vol. 32, No.2, pp. 121-213, 2003
4. Гайле А.А., Сомов В.Е., Варшавский О.М. «Ароматические углеводороды: Выделение, применение, рынок»: Справочник. - СПб.: Химиздат, 2000г. 12с.
5. Гайле А.А., Сомов В.Е., Варшавский О.М., Семенов Л.В. «Сульфолан: свойства и применение в качестве селективного растворителя», Спб.: Химииздат, 1998г. 81 с.
6. Петлюк Ф. Б., Серафимов Л. А. Многокомпонентная ректификация: теория и расчет. М.:Химия, 1983.
7. Александров И. А. Ректификационные и абсорбционные аппараты. М.:Химия, 1978.
8. Общий курс процессов и аппаратов химической технологии. Книга 2. под ред. В. Г. Айнштейна. Москва. Химия,2000.
9. Айнштейн В.Г., Захаров М.К., Носов Г.А., Захаренко В.В., Зиновкина Т.В., Таран А.Л., Костаян А.Е. «Общий курс процессов и аппаратов химической технологии» т.2 Москва «Логос» «Высшая школа» 2003 г. 1758с.
10. Львов С.В. Некоторые вопросы ректификации бинарных и многокомпонентных смесей, изд. АНСССР, 1960 г, 163 с.
11. Серафимов Л. А., Мозжухин А.С., Науменкова Л. Б. Определение числа вариантов технологических схем ректификации n-компонентных зеотропных смесей, ТОХТ, т. 27, N3, с. 292-295,1993
12. Морачевский А.Г., Смирнова Н.А., Пиотровская Е.М. и др. “Термодинамика равновесия жидкость–пар”, под ред. Морачевского А.Г. – Л.: Изд–во Химия, 1989, с. 344.
13. Nitta T., Katayama T.//J. Chem. Eng. Jap. 1974, v.7 p
381-382]
14. Hiranuma N.// Ind. Eng. Chem. Fund. 1974 V.13.,p.219-222
15. Palmen D. A., Smith D. B.// Ind. Eng.Chem., Proc. Des. Dev. 1972. V.1. p
114-119.
|