А.А. Замятнин, Институт биохимии им. А.Н. Баха РАН, г. Москва
Увидев в заголовке статьи термин «слабые взаимодействия», возможно, кто-то подумает, что речь пойдет об элементарных частицах. Не знаю, разочарую я их или нет, но далее никакого упоминания об элементарных частицах не будет.
Дело в том, что термин «слабые взаимодействия» (как, впрочем, и сильные) в результате несоблюдения семантической гигиены (термин Н.В. Тимофеева-Ресовского) используется не только в физике элементарных частиц. В частности, эти взаимодействия характерны для огромного числа органических веществ, среди которых особое положение занимают вещества биологической природы – белки, нуклеиновые кислоты, углеводы, липиды и многие другие. Их структура определяется двумя факторами.
Атомы в молекулах этих веществ соединены ковалентными связями (сильные взаимодействия), формирующими химическую структуру. Однако реальная конфигурация зависит от наличия у них разнообразных химических групп, которые внутри молекулы на основании законов физики могут специфически взаимодействовать друг с другом (слабые взаимодействия) и формировать уникальную пространственную структуру. Более того, возникающая при этом система слабых внутримолекулярных взаимодействий в силу ряда причин может изменяться, перестраиваться, в результате чего молекула приобретает разнообразные формы и наделяется способностью участвовать еще и во множестве слабых межмолекулярных взаимодействий.
Межмолекулярные взаимодействия являются первичным актом многоэтапного процесса, который заканчивается макроскопическими изменениями живого организма. Сложность и многообразие системы слабых взаимодействий в живой природе требует от исследователя использования разнообразных методов и подходов, большинство из которых на сегодняшний день основаны на высоких технологиях.
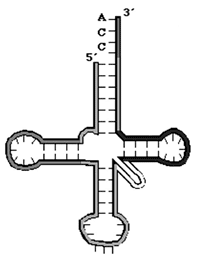
«Клеверный лист» молекулы транспортной РНК образуется за счет внутримолекулярных слабых взаимодействий (водородных связей) между азотистыми основаниями
|
Наиболее ярко продемонстрировать слабые взаимодействия можно на примере белков, для которых из-за отсутствия теоретической базы до сих пор не решена фундаментальная задача формирования пространственной структуры и не ясны механизмы их взаимодействия с многими другими молекулами.
Реклама

На сегодняшний день известно около 2 млн белков с разной химической структурой, синтезируемых особыми структурными элементами живой клетки – рибосомами. Молекулы всех белков обладают общим свойством: их характеризует линейная последовательность повторяющихся трех химических групп, соединенных ковалентными связями, которые представляют собой остов белковой молекулы. Вокруг этих связей может происходить вращение. К одной из трех групп (всегда одной и той же) присоединен специфический боковой радикал (один из 20 возможных). Участок остова, состоящий из трех химических групп с присоединенным боковым радикалом, представляет собой аминокислотный остаток. В процессе синтеза белка на рибосоме аминокислотные остатки образуются из аминокислот и занимают свое положение в белке, соединяясь друг с другом специфической ковалентной связью, называемой пептидной. Поэтому все соединения такого типа называются пептидами. Число аминокислотных остатков в одной молекуле может превышать 30 тыс.
 |
Слабые взаимодействия, например водородные связи, между химическим группами остова во многих белках приводят к образованию в них регулярных участков, представляющих собой спирали или слоистые структуры. Однако уникальная пространственная структура образуется с участием и ряда других слабых взаимодействий между боковыми радикалами. Большое их число (несмотря на слабость) обусловливает то, что белковая молекула может обладать стабильной пространственной конфигурацией.
Среди 20 видов боковых радикалов имеются заряженные (положительно или отрицательно), гидрофобные, гидрофильные, содержащие циклические группы и др. Их сближение характеризуется целым спектром слабых взаимодействий: ионных, ион-дипольных, диполь-дипольных (разнообразных вандерваальсовских – совокупность ориентационных, индукционных и дисперсионных сил), гидрофобных и др. Энергия U слабых взаимодействий в разной степени n зависит от расстояния r между взаимодействующими элементами:
U ~ 1/rn.
Для сравнения в таблице представлены характеристики различных типов слабых взаимодействий и ковалентных связей (сильные взаимодействия) в белках.
| Тип взаимодействий |
n |
Энергия, кДж/моль |
| Ковалентные связи |
– |
>300 |
| Ионные взаимодействия |
1 |
<100 |
| Ион-дипольные взаимодействия |
2 |
15–40 |
Диполь-дипольные взаимодействия
(вандерваальсовские)
|
4, 6 |
4–100 |
| Водородные связи |
2 |
10–30 |
Особое место в формировании структуры белка принадлежит гидрофобным взаимодействиям, которые возникают из-за отталкивания молекул воды неполярными боковыми группами. В результате часто образуется гидрофобное ядро молекулы.
Реклама

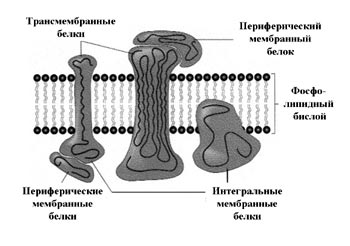
Схематическое изображение биологического устройства биологической мембраны.
Все ее компоненты связаны между собой слабыми взаимодействиями
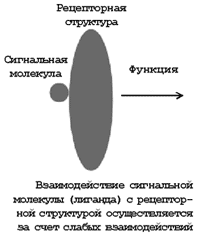
Макроскопический физиологический процесс может проявиться в результате слабых взаимодействий даже в одной (!) или нескольких парах сблизившихся молекул. Например, при взаимодействии пептидной молекулы с белковым рецептором нервной системы происходит сокращение мышцы. При этом осуществляются слабые взаимодействия в нескольких контактах, т.е. в соответствии с таблицей энергия суммарного взаимодействия, например 5 контактов, составит величину порядка 105 Дж/моль или (с учетом числа Авогадро) ~10–19 Дж на одну взаимодействующую пару молекул. Как же мала эта величина по сравнению с 10 Дж, характеризующими работу, совершаемую человеком при поднятии 1 кг груза на высоту 1 м!
Сегодня при наличии сравнительно несложного и недорогого оборудования расшифровка химических структур не представляет труда и за эту работу не присуждают Нобелевских премий, как это было всего полвека назад. Иначе обстоит дело с пространственной структурой. Ее определяют, используя два мощных и дорогостоящих физических метода – рентгеноструктурный анализ (РСА) и ядерный магнитный резонанс (ЯМР). С помощью РСА впервые были определены пространственные структуры достаточно больших белковых молекул – миоглобина и гемоглобина, и за эту работу авторы были удостоены Нобелевской премии. Сейчас с применением этого метода установлена пространственная структура уже нескольких тысяч белков. С помощью ЯМР ежегодно определяются десятки структур сравнительно небольших молекул.
Несмотря на явный прогресс, расшифровка пространственных структур остается весьма трудоемким и дорогостоящим процессом. Так, определение структуры лишь одного белка методом РСА требует работы большого коллектива в течение почти года и стоит около 300 тыс. долларов. Можно себе представить, сколько времени и денег понадобится для определения пространственных структур миллионов белков, если следовать этим путем.
Очевидно, возникает вопрос: если природа из множества возможных пространственных структур белка однозначно формирует только одну, то, видимо, это определяется уникальной последовательностью его аминокислотных остатков? А может быть, зная эту последовательность, используя существующие на сегодняшний день высокие технологии, можно смоделировать процесс сворачивания и получить пространственную структуру без огромных затрат времени и денег? Такой вопрос возник уже около 40 лет назад, и многие физики, осознав важность проблемы, занялись ее решением, потратив на это большую часть свого времени, а некоторые даже и всю жизнь. Однако проблема так и остается нерешенной. Почему?
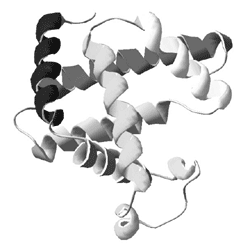
Альфа-спирали в белках тоже образуются за счет внутримолекулярных водородных связей
|
Причин этому несколько. Физики долгое время рассматривали молекулы белков как статистический ансамбль, который может быть подвергнут анализу методами теоретической физики. Они брали последовательность аминокислотных остатков белка и, исходя из того, что данная система должна стремиться к термодинамическому равновесию, методами молекулярного моделирования сворачивали линейную молекулу белка, отыскивая конфигурацию с минимальной энергией, которую полагали соответствующей пространственной структуре. В этом случае моделирование начиналось с уже полностью приготовленной белковой молекулы.
Но даже из энергетических соображений невозможно себе представить, чтобы большая молекула белка сначала полностью образовалась в виде линейной структуры, а только потом начала бы сворачиваться. Да и в природе все происходит иначе. Линейная последовательность аминокислотных остатков выходит из синтезирующего аппарата рибосомы всегда одним и тем же (из двух возможных) концом и, шаг за шагом удлиняясь, приобретает определенную форму. Это сворачивание, сопровождающее трансляцию, называется ко-трансляционным фолдингом.
Более того, рассматривавшиеся физиками белки обычно были только частью аминокислотной последовательности, синтезированной на рибосоме. Полная последовательность в своей начальной области может содержать короткие, так называемые пре- и про-участки. Эти участки впоследствии отщепляются подобно ступеням космических аппаратов (но в обратном порядке), и только тогда оставшаяся часть становится функциональным белком. Поэтому возникает вопрос: идентична ли структура самого белка структуре его предшественника? Как недавно выяснилось, белок может приобрести совершенно другую конфигурацию после удаления даже единичных концевых аминокислотных остатков.
Существуют и другие причины, которые усугубляют трудности, связанные с теоретическим выявлением пространственной структуры белка. К ним относится учет влияния молекул воды, ионов и других низко- и высокомолекулярных соединений. Как оказалось, в сворачивании белка также принимают участие и специализированные молекулы (шапероны), предотвращающие агрегацию.
Безуспешные попытки решения проблемы фолдинга связаны, как ни странно, и с использованием высоких технологий, в частности с применением сложных компьютерных программ. Сразу же отметим, что программ, правильно моделирующих ко-трансляционный фолдинг, на сегодняшний день просто не существует. Но даже в те из них, которые позволяют оптимизировать структуру и сводить ее к минимальному энергетическому состоянию, введены численные параметры разных взаимодействий, которые определены, разумеется, с некоторой ошибкой. При большом числе шагов оптимизации структуры (1000 и более) эта ошибка вырастает до таких величин, которые позволяют усомниться в правильности полученного результата.
Для стимулирования прогресса в решении этой проблемы в г. Асиломар (США) уже несколько лет проводится конкурс на лучшее определение пространственной структуры белка по его аминокислотной последовательности. Для этого организаторы конкурса выбирают еще неопубликованную работу, в которой методом РСА выявлена пространственная структура нового белка и, сообщив каждому желающему лишь аминокислотную последовательность, предлагают предсказать структуру любым теоретическим методом. Желающих бывает до нескольких десятков, они делают свои предсказания, которые организаторы сравнивают с имеющейся в их распоряжении структурой и выявляют победителей конкурса. Однако никто (в том числе и победители) никогда не представил структуры, полностью соответствующей той, что получена экспериментально.
Складывается впечатление, что выбранные пути непригодны для решения этой проблемы. Вероятно, нужны принципиально новые идеи для того, чтобы произошел прорыв в данной области. Впрочем, одна такая идея существует и уже давно, хотя до настоящего времени ее не признает большинство компетентных физиков и биологов. В чем же она состоит?
Около 30 лет назад у нас в стране Л.Б. Меклер и Р.Г. Идлис предложили механизм фолдинга, который основан на чисто формальном постулате. Он состоит в том, что в формирующемся предшественнике белка пространственно сближаются пары аминокислотных остатков, которые в нуклеиновых кислотах кодируются кодоном и соответствующим антикодоном. Сразу заметим, что для этой идеи нет не только научного обоснования, но в ней полностью отсутствует и какой-либо учет физических взаимодействий (т.е. идея, которую вполне можно отнести к разряду сумасшедших). Однако авторы учитывают и однонаправленность синтеза, и то, что вначале образуется предшественник, а не сам белок. В процессе такого ко-трансляционного фолдинга каждый новый присоединенный аминокислотный остаток сопоставляется с введенными ранее, и если в соответствии с постулатом он находит себе партнера, то располагается рядом с ним, образуя некие необъясненные связи.
Вместе с квалифицированными специалистами-структурщиками в начале 1990-х гг. мне довелось участвовать в проверке работоспособности этого механизма, и, что удивительно, попытки построения нескольких десятков структур каждый раз приводили к точному результату. Более того, однажды была взята аминокислотная последовательность, в которой, как оказалось позднее, всего лишь один остаток ошибочно не соответствовал правильному. В этом случае должной структуры не получилось, но после исправления ошибки все стало на свое место.
Вскоре авторы переехали в Израиль, где было намечено создать компьютерную программу, воспроизводящую их процесс фолдинга. Однако из-за кончины Л.Б. Меклера и отсутствия соответствующего финансирования эта работа приостановилась. А жаль!
Пространственные структуры некоторых белков, полученные по правилам Меклера и опубликованные на обложке журнала «Природа» в 1993 году
Подведем итог. На сегодняшний день мы немало знаем о слабых взаимодействиях в белках, но эти знания не помогли нам раскрыть тайну фолдинга (возможно, существуют такие типы взаимодействий, которые пока еще не выявлены). И хотя существует успешно применяемый теоретический метод, но в нем отсутствует понимание природы возникновения взаимодействий, а оно, разумеется, должно быть. Наверное, истина, как всегда, находится где-то посередине, и современные высокие технологии должны ее выявить.
Может быть, понадобятся и новые идеи (включая сумасшедшие). Но в любом случае без высоких технологий не обойтись, как бы дорого они ни стоили. Например, затраты на создание компьютерной программы для изучения любого белка на основе механизма Меклера–Идлис оцениваются примерно в 100 тыс. долл., но разве это много по сравнению с затратами на выявление структуры всего лишь одного белка методом РСА? И как было бы замечательно, если бы успешное решение застарелой проблемы осуществилось в нашей стране.
Список литературы
1. Замятнин А.А. Блистающий мир белков и пептидов / Биология. 2002. № 25–26. С. 8–13.
2. Замятнин А.А. Протеомика / Биология. 2005. № 14. С. 2–11.
3. Меклер Л.Б., Идлис Р.Г. Общий стереохимический генетический код – путь к биотехнологии и универсальной медицине XXI в. уже сегодня. Коммент. В.Т. Иванова, Д.Г. Кнорре, М.А. Мокульского, А.А. Замятнина /Природа. 1993. № 5. С. 28–70.
|
