КОНТРОЛЬНАЯ РАБОТА ПО ПРЕДМЕТУ:
ФИЗИЧЕСКАЯ И КОЛЛОИДНАЯ ХИМИЯ
Содержание
1. Влияние температуры на скорость химических процессов. Правило Вант-Гоффа
2. Второй закон термодинамики. Самопроизвольные процессы. Свободная и связанная энергия. Энтропия
3. Зависимость скорости химической реакции от концентрации реагирующих веществ. Закон действующих масс
3.1 На основании закона действующих масс запишите формулы скорости для химических реакций
4. Давление пара над растворами. Первый закон Рауля
5. Зависимость адсорбции от свойств твердой поверхности. Гидрофильные и гидрофобные поверхности
5.1 Выбрать правильный ответ и пояснить их
6. Вопрос. Пищевые пены: понятия, виды, их состав и строение, влияние на консистенцию пищи
Список использованных источников
Скорость химической реакции - это величина, показывающая как изменяются концентрации исходных веществ или продуктов реакции за единицу времени.
Скорость химической реакции зависит от природы реагирующих веществ и условий протекания реакции: концентрации с, температуры t, присутствия катализаторов, а также от некоторых других факторов (например, от давления - для газовых реакций, от измельчения - для твердых веществ, от радиоактивного облучения).
Влияние концентраций реагирующих веществ. Чтобы осуществля¬лось химическое взаимодействие веществ А и В, их молекулы (частицы) должны столкнуться. Чем больше столкновений, тем быстрее протекает реакция. Число же столкновений тем больше, чем выше концентрация реагирующих веществ. Отсюда на основе обширного экспериментального материала сформулирован основной закон химической кинетики, устанавливающий зависимость скорости реакции от концентрации реагирующих веществ:
Скорость химической реакции пропорциональна произведению концентраций реагирующих веществ.
Для реакции (I) этот закон выразится уравнением
v = kcA
cB
, (1)
где сА и сВ - концентрации веществ А и В, моль/л; k - коэффициент пропорциональности, называемый константой скорости реакции. Основной закон химической кинетики часто называют законом действующих масс.
Из уравнения (1) нетрудно установить физический смысл константы скорости k: она численно равна скорости реакции, когда концентрации каждого из реагирующих веществ составляют 1 моль/л или когда их произведение равно единице.
Реклама

Константа скорости реакции k зависит от природы реагирующих веществ и от температуры, но не зависит от их концентраций.
Уравнение (1), связывающее скорость реакции с концентрацией реагирующих веществ, называется кинетическим уравнением реакции. Если опытным путем определено кинетическое уравнение реакции, то с его помощью можно вычислять скорости при других концентрациях тех же реагирующих веществ.
Зависимость скорости реакции от температуры определяется правилом Вант-Гоффа:
При повышении температуры на каждые 10о скорость большинства реакций увеличивается в 2-4 раза.
Математически эта зависимость выражается соотношением 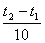
vt 2
= vt 1
γ,
где vt 1, vt 2 - скорости реакции соответственно при начальной (t 1) и конечной (t 2) температурах, а γ - температурный коэффициент скорости реакции, который показывает, во сколько раз увеличивается скорость реакции с повышением температуры реагирующих веществ на 10°.
Правило Вант-Гоффа является приближенным и применимо лишь для ориентировочной оценки влияния температуры на скорость реакции. Температура влияет на скорость химической реакции, увеличивая константу скорости.
Вычислите, во сколько раз уменьшится скорость реакции, протекающей в газовой фазе, если понизить температуру от 120º до 60ºС. Температурный коэффициент равен 3.
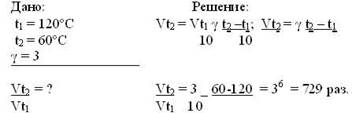
Ответ. Скорость химической реакции при понижении температуры от 120º до 60º С уменьшится в 729 раз.
Химическая термодинамика - наука, изучающая условия устойчивости систем и законы.
Термодинамика - наука о макросистемах (рис.1).

Рисунок 1. Схема составляющих термодинамической науки
Первое начало термодинамики утверждает, что при превращении одной формы энергии в другую полная энергия системы не изменяется, однако не указывает никаких ограничений относительно возможности этого процесса. Поэтому первое начало термодинамики позволяет рассчитать энергетический эффект процесса, однако не дает ответа на вопросы о том, будет ли процесс протекать самопроизвольно, о направлении и глубине протекания процесса.
Самопроизвольный процесс - процесс, который может протекать без затраты работы извне, причем в результате может быть получена работа в количестве, пропорциональном произошедшему изменению состояния системы. Самопроизвольный процесс может протекать или обратимо, или необратимо. Чтобы самопроизвольный процесс протекал обратимо, необходимо приложить извне такое сопротивление, чтобы переход был очень медленным и при бесконечно малом изменении противодействующей силы процесс мог пойти в обратном направлении. В случае обратимо происходящего изменения состояния системы производится максимальное количество работы. Всякий реальный процесс в какой-то степени является необратимым, и получаемая работа меньше максимально возможного теоретического количества.
Реклама

Вынужденный процесс - процесс, для протекания которого требуется затрата работы извне в количестве, пропорциональном производимому изменению состояния системы.
Второе начало термодинамики дает возможность определить, какой из процессов будет протекать самопроизвольно, какое количество работы может быть при этом получено, каков предел самопроизвольного течения процесса. Далее, второе начало термодинамики дает возможность определить, какими должны быть условия, чтобы нужный процесс протекал в необходимом направлении и в требуемой степени, что особенно важно для решения различных задач прикладного характера.
Естественные процессы всегда направлены в сторону достижения оси с темой равновесного состояния (механического, термического или любого другого). Это явление отражено вторым законом термодинамики, имеющим большое значение и для анализа работы теплоэнергетических машин. В соответствии с этим законом, например, теплота самопроизвольно может переходить только от тела с большей температурой к телу с меньшей температурой. Для осуществления обратного процесса должна быть затрачена определенная работа. В связи с этим второй закон термодинамики можно сформулировать следующим образом: невозможен процесс, при котором теплота переходила бы самопроизвольно от тел более холодных к телам более теплым
(постулат Клаузиуса, 1850 г.).
Любая форма энергии может полностью перейти в теплоту, но теплота преобразуется в другие формы энергии лишь частично.
химический термодинамика скорость раствор
Таким образом, можно условно принять, что внутренняя энергии системы состоит из двух составляющих: "свободной" X и "связанной" Y энергий, причем "свободная" энергия может быть переведена в работу, а "связанная" энергия может перейти только в теплоту.
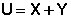
Мерой связанной энергии является новая термодинамическая функция состояния, называемая энтропией.
В природе протекание большинства процессов, в том числе и химических, сопровождается не только энергетическими эффектами, но и изменением в упорядочении расположения частиц относительно друг друга. Рассмотренные выше примеры превращений имеют одно общее свойство: в каждом случае состояние продуктов характеризуется большей хаотичностью, или неупорядоченностью, чем состояние реагентов. Растворение хлорида калия сопровождается нарушением регулярности в расположении частиц в узлах кристаллической решетки - возникает беспорядочное распределение ионов в растворе. Молекулы воды, образующие кристалл льда, прочно удерживаются в его кристаллической решетке. При плавлении льда молекулы H2O начинают свободно перемещаться относительно друг друга. Высокоупорядоченная кристаллическая структура заменяется неупорядоченной структурой жидкости. В процессе испарения структура жидкости, представленная ассоциатами из ее молекул, заменяется отдельными молекулами, движущимися независимо (в газовой фазе).
Таким образом, частицам (молекулам, атомам, ионам и др.) присуще стремление к беспорядочному движению, поэтому система стремится перейти из более упорядоченного состояния в менее упорядоченное. Количественной мерой неупорядоченности (беспорядка) системы является термодинамическая функция состояния системы - энтропия (S, Дж/ (мольK)). Чем в большей мере выражен беспорядок в системе, тем больше ее энтропия. Следовательно, еще одной составляющей движущей силы самопроизвольно протекающих процессов является тенденция к увеличению энтропии системы.
II закон термодинамики является одним из наиболее общих положений всей науки в целом. Главная мысль его заключается в том, что в любой изолированной системе с течением времени происходит постоянное возрастание степени беспорядка, т.е. энтропии. Следовательно, для любых самопроизвольных процессов
ΔS ≥ 0.
Знак ">" - для необратимых процессов, знак "=" - для обратимых процессов.
Для обратимых процессов ΔS = Q/T, [Дж/К моль].
Для необратимых процессов ΔS > Q/T, [Дж/К моль].
II закон термодинамики имеет ясный физический смысл только тогда, когда его применяют к любой ограниченной системе. Функции системы, которые связаны с работой и говорящие о направлении процесса, называются термодинамическими потенциалами. Критерием для суждения о направлении процессов в изолированных системах может служить изменение энтропии ΔS. Однако на практике большинство процессов протекает в неизолированных системах и связано с теплообменом и изменением объема. Поэтому для неизолированных систем необходимо иметь другие критерии. Энтропию веществ принято относить к стандартным условиям (T = 298,15 K и p = 101,3 кПа). Энтропию при этих условиях называют стандартной энтропией и обозначают S° (298 K). Значения стандартных энтропий для многих веществ являются справочными данными.
2.1 Объясните изменение энтропии в процессах
Уравнение реакции позволяет судить о знаке изменения энтропии ∆S0
.
а) 3Н2 (
г) + N2
(г) = 2 NН3
(г)
В реакции число молей газообразных веществ уменьшается от 4 до 2, поэтому ∆S0
< 0
б) С (тв) + Н2
О (г) = СО (г) + Н2
(г).
Из реакции следует, что из 1 моля твердого С и 1 моль газообразного Н2
Oобразуется 2 моль газообразных веществ (1 моль СО (г) и 1 моль Н2
(г)). Следовательно ∆S0
> 0.
Химические реакции протекают с различными скоростями. Некоторые из них полностью заканчиваются за малые доли секунды (взрыв), другие осуществляются за минуты, часы, дни и большие промежутки времени. Кроме того, одна и та же реакция может в одних условиях (например, при повышенных температурах) протекать быстро, а в других (например, при охлаждении) - медленно. При этом различие в скорости одной и той же реакции может быть очень большим.
Раздел химии, изучающий скорости химических реакций, называется химической кинетикой.
При рассмотрении вопроса о скорости реакций необходимо различать гомогенные и гетерогенные реакции. С этими понятиями тесно связано понятие фазы.
Фазой называется часть системы, отделенная от других ее частей поверхностью раздела, при переходе через которую свойства изменяются скачком.
Гомогенная реакция протекает в объеме фазы [пример - взаимодействие водорода и кислорода с образованием водяного пара: H2 (г) + O2 (г) → H2O (г)], а если реакция гетерогенна, то она протекает на поверхности раздела фаз [например, горение углерода: C (т) + O2 (г) → CO2 (г)]. Скоростью гомогенной реакции называется количество вещества, вступающего в реакцию или образующегося при реакции за единицу времени в единице объема фазы:
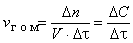 , ,
где n - количество вещества, моль; V - объем фазы, л; t - время; С - концентрация, моль/л.
Скоростью гетерогенной реакции называется количество вещества, вступающего в реакцию или образующегося при реакции за единицу времени на единице площади поверхности фазы:
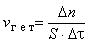 , ,
где S - площадь поверхности раздела фаз.
К важнейшим факторам, влияющим на скорость гомогенной реакции, являются следующие: природа реагирующих веществ, их концентрации, температура, присутствие катализаторов.
Зависимость скорости реакции от концентраций реагирующих веществ. Реакция между молекулами происходит при их столкновении. Поэтому скорость реакции пропорциональна числу соударений, которые претерпевают молекулы реагирующих веществ. Число соударений тем больше, чем выше концентрация каждого из исходных веществ. Например, скорость реакции A + B → C пропорциональна произведению концентраций А и В:
v = k× [A] × [B],
где k - коэффициент пропорциональности, называемый константой скорости реакции. По смыслу величина k равна скорости реакции для случая, когда концентрации реагирующих веществ равны 1 моль/л.
Это соотношение выражает закон действия масс. Этот закон называют также законом действующих масс.: при постоянной температуре скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ.
Гораздо реже реакция осуществляется в результате одновременного столкновения трех реагирующих частиц. Например, реакция
2А+В → А2В
может протекать путем тройного столкновения:
А+А+В → А2В
Тогда в соответствии с законом действия масс концентрация каждого из реагирующих веществ входит в выражение скорости реакции в степени, равной коэффициенту в уравнении реакции:
v = k× [A] × [A] × [B] = k× [A] 2 [B]
Сумма показателей степени в уравнении закона действия масс называется порядком реакции. Например, в последнем случае реакция имеет третий порядок (второй - по веществу A и первый - по веществу B).
В случае, когда уравнение химической реакции не соответствует элементарному акту взаимодействия, а отражает лишь связь между массой вступивших в реакцию и образовавшихся веществ, то степени у концентраций не будут равны коэффициентам, стоящим перед формулами соответствующих веществ в уравнении реакции. Для реакции, которая протекает в несколько стадий, скорость реакции определяется скоростью самой медленной (лимитирующей) стадии.
Такая зависимость скорости реакции от концентрации реагирующих веществ справедлива для газов и реакций, проходящих в растворе. Реакции с участием твердых веществ не подчиняются закону действующих масс, так как взаимодействие молекул происходит лишь на поверхности раздела фаз. Следовательно, скорость гетерогенной реакции зависит еще и от величины и характера поверхности соприкосновения реагирующих фаз. Чем больше поверхность - тем быстрее будет идти реакция.
а) СО (г) + Сl2
(г) → СОСl2
(г);
v= k [CO] [Cl2
]
б) С12
Н22
O11
(водн.) + Н2
O (ж) → 2С6
Н12
О6
(водн);
v= k
в) С (тв) + О2
(г) → СО2
(г)
v= k [O2
]
Растворы - это гомогенные смеси, состоящие из нескольких компонентов (минимум из двух), каждый из которых распределен по всему объему раствора в виде отдельных атомов, молекул, ионов или в виде групп из небольшого числа этих частиц. Состав растворов может непрерывно меняться в границах, определяемых взаимной растворимостью веществ, т.е. растворы не подчиняются закону постоянства состава. Таким образом, в этих определениях подчеркиваются два основных признака растворов (и электролитов, и неэлектролитов): их гомогенность и переменный состав.
От химических соединений растворы отличают характер и величины энергии связи между частицами растворенного вещества и растворителя. Если химические соединения образуются за счет валентных связей атомов, то растворы образуются за счет слабых межмолекулярных (Ван-дер-Ваальсовых) сил. Эти силы действуют как между молекулами одного и того же компонента, так и между молекулами разных компонентов. Существенное значение могут иметь и водородные связи.
Общими признаками растворов являются самопроизвольность образования их при постоянных температуре и давлении и устойчивость их как термодинамических систем (ΔG < 0).
Растворы могут существовать в жидком, твердом, газообразном состояниях.
Число компонентов в растворе не может быть меньше двух. Компонент, содержащийся в большем количестве, называется растворителем, а в меньшем - растворенным веществом.
Истинные растворы делятся на идеальные разбавленные растворы, идеальные концентрированные и неидеальные (реальные) растворы.
Если для газа условием идеальности является отсутствие сил взаимодействия между молекулами, то для раствора условием идеальности является однообразие силы взаимодействия между молекулами компонентов, образующих раствор.
К идеальным разбавленным растворам относятся растворы с концентрацией меньше 1 моль на 1000 г растворителя.
Идеальные концентрированные растворы образуют вещества, близкие по физическим и химическим свойствам; между молекулами компонентов не происходит каких-либо взаимодействий химического характера. Образование таких растворов не сопровождается тепловыми эффектами или изменением объема. Как правило, свойствами идеальных концентрированных растворов обладают растворы изотопов данного элемента, смеси гомологов, оптических изомеров.
Реальными называются растворы, которые не подчиняются законам идеальных растворов. Обычно они составлены из компонентов с различными свойствами, строением молекул и силами взаимодействия между молекулами компонентов. Образование таких растворов сопровождается изменением объема и тепловыми эффектами. С уменьшением концентрации реального раствора его свойства приближаются к свойствам идеального раствора.
В жидких растворах частицы растворенного вещества связаны с окружающими их частицами растворителя. Эти комплексы называются сольватами, а для водных растворов - гидратами. Именно такое представление о растворах было высказано Д.И. Менделеевым. На основании экспериментальных фактов он выдвинул предположение о существовании в растворах определенных химических соединений растворенного вещества с растворителем. Эта идея составила основу химической теории растворов: образование раствора - химический процесс взаимодействия молекул растворенного вещества и растворителя.
Физическая теория, существовавшая ранее, рассматривала растворы как простые механические смеси, а растворитель как инертную среду.
Закономерности для разбавленных молекулярных растворов были выведены в 80-х годах 19 века Ф. Раулем, Я. Вант-Гоффом, С. Аррениусом, которые установили зависимости между концентрацией раствора и такими его свойствами как давление насыщенного пара, температуры кипения и замерзания, осмотическое давление.
1) Давление насыщенного пара является важным свойством раствора.
Если к чистому растворителю с давлением насыщенного пара р0 добавить постороннее нелетучее вещество, то давление пара растворителя над раствором изменится.
При растворении какого-либо нелетучего вещества в данном растворителе понижается концентрация молекул последнего в единице объема жидкости, и уменьшается число молекул, вылетающих в единицу времени из жидкой фазы в парообразную. При меньшей концентрации пара, т.е. при меньшем его давлении, установится равновесие.
Следовательно, давление насыщенного пара растворителя над раствором всегда меньше, чем над чистым растворителем. Понижение давления насыщенного пара растворителя над раствором тем больше, чем выше концентрация растворенного вещества.
Для идеальных растворов давление насыщенного пара определяется законом Рауля, установленного им в 1886 г.: относительное понижение давления насыщенного пара растворителя над раствором равно молярной доле растворенного вещества, т.е. отношению количества данного вещества к общему количеству растворителя и растворенного вещества:
Δр / р0 = р0 - р/р0 = n2/n1 + n2 = х2, где
р0 - давление пара растворителя над чистым растворителем, р - давление пара растворителя над раствором, Δр / р0 - относительное понижение давления пара растворителя, n2 - количество растворенного вещества, n1 - количество вещества растворителя, х2 - молярная доля растворенного вещества.
Это уравнение можно представить в другом виде и придать закону иную формулировку:
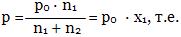
давление пара над раствором равно произведению давления пара над чистым растворителем на молярную долю растворителя.
Для очень разбавленных растворов уравнение имеет вид:
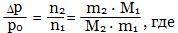
m1 и m2 - массы растворителя и растворенного вещества соответственно, М1 и М2 - молярные массы растворителя и растворенного вещества соответственно.
2) Понижение давления насыщенного пара влечет за собой понижение температуры замерзания раствора по сравнению с чистым растворителем.
Жидкость замерзает при той температуре, при которой давление насыщенного пара над ней такое же, как и над кристаллами этого вещества. Так как давление насыщенного пара растворителя над раствором всегда меньше, чем над чистым растворителем, то разбавленный раствор будет замерзать при более низкой температуре, чем растворитель.
Температурой замерзания раствора считают ту температуру, при которой в процессе охлаждения начинают выделяться первые кристаллы чистого растворителя.
Для таких растворов Рауль нашел, что понижение температуры замерзания раствора Δtз. = t0 - t (t0 - температура замерзания растворителя, t - температура замерзания раствора) пропорционально его моляльности (1 моль в 1000 г растворителя):
Δtз = К m,
где
Δt - понижение температуры замерзания, m - моляльность раствора, К - криоскопическая постоянная ("криос" - холод).
Физический смысл криоскопической постоянной К состоит в том, что она равна понижению tз. раствора, содержащего 1 моль растворенного вещества на 1 кг растворителя. Величина К зависит от природы растворенного вещества, если его молекулы не диссоциируют и не ассоциируют. Величину К можно рассчитать по формуле
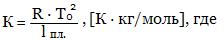
T0 - температура замерзания растворителя, l пл - удельная теплота плавления растворителя.
Так как 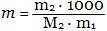 [моль/кг], то [моль/кг], то
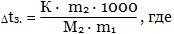
m1 - масса растворителя, m2 - масса растворенного вещества.
3) Повышение температуры кипения.
Жидкость закипает при температуре, при которой давление насыщенного пара жидкости становится равным внешнему давлению. Так как давление насыщенного пара растворов нелетучих или малолетучих веществ меньше давления насыщенного пара растворителя, то эти растворы кипят при более высокой температуре, чем растворитель.
Для разбавленных растворов таких веществ Рауль установил, что повышение температуры кипения раствора Δtк. = t - t0 пропорционально его моляльности:
Δtк. = Е · m, где
Е - эбуллиоскопическая постоянная ("эбуллиос" - кипеть), m - моляльность раствора.
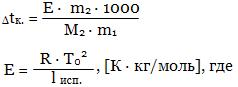
T0 - температура кипения растворителя, lисп. - удельная теплота испарения растворителя.
Величина Е численно равна повышению tк моляльного раствора при условии сохранения свойств раствора до этой концентрации.
В 100 г. этилового спирта растворено 39г. бензола С6
Н6
, рассчитайте повышение температуры кипения этого раствора, если эбуллиоскопическая константа спирта равна 1,11ºС.
Дано:

Ответ: ∆tº кип. = 5.55 ºС
Адсорбция (от лат. ad - на, при и sorbeo - поглощаю), поглощение вещества из газообразной среды или раствора поверхностным слоем жидкости или твёрдого тела. Например, если поместить в водный раствор уксусной кислоты кусочек угля, то произойдёт адсорбция - количество кислоты в растворе уменьшится, молекулы кислоты сконцентрируются на поверхности угля.
Вещество, на поверхности которого происходит адсорбция, называется адсорбентом, а поглощаемое из объёмной фазы - адсорбатом. В зависимости от характера взаимодействия между молекулой адсорбата и адсорбентом адсорбцию принято подразделять на физическую и хемосорбцию. Менее прочная физическая адсорбция не сопровождается существенными изменениями молекул адсорбата. Она обусловлена силами межмолекулярного взаимодействия, которые связывают молекулы в жидкостях и некоторых кристаллах и проявляются в поведении сильно сжатых газов. При хемосорбции молекулы адсорбата и адсорбента образуют химические соединения. Часто адсорбция обусловлена и физическими и химическими силами, поэтому не существует чёткой границы между физикой адсорбцией и хемосорбцией.
Поверхность твердого тела, в отличие от поверхности жидкости, имеет сложный, неоднородный характер. Даже полированное зеркало имеет на поверхности выступы размерами до  см. Адсорбция происходит не на всей поверхности, а лишь на активных центрах. см. Адсорбция происходит не на всей поверхности, а лишь на активных центрах.
Поверхность адсорбента часто бывает пористой. Наличие пор приводит к тому, что адсорбция сопровождается капиллярной конденсацией.
Величина адсорбции газа на твердом адсорбенте зависит от следующих факторов:
температуры;
концентрации (равновесного давления) пара или газа в поверхностном слое;
природы твердого тела;
природы газа.
Особенности адсорбции газов на твердых адсорбентах свидетельствуют о том, что определяющим в этом виде адсорбции является состояние поверхности адсорбента.
Все адсорбенты подразделяются на две принципиально разные группы:
1) адсорбенты с гладкой поверхностью (непористые адсорбенты);
2) пористые адсорбенты.
При одинаковой степени измельчения пористые адсорбенты имеют гораздо большую удельную поверхность чем непористые, и, кроме того, адсорбция на них может сопровождаться капиллярной конденсацией.
Адсобрция на непористых адсорбентах зависит в основном:
от сродства адсорбента к адсорбтиву. Это сродство тем сильнее, чем резче выражена склонность к образованию определенных связей. Так, графитированная сажа неполярна, поэтому на ней сильнее адсорбируются неполярные органические соединения. На поверхности ионных кристаллов (полярные адсорбенты) лучше адсорбируются полярные вещества. На поверхности оксидов обычно имеются гидроксильные группы, способные образовывать водородные связи, поэтому они прочно удерживают воду, спирты, амины и т.д.;
от дисперсности адсорбента. Вы помните, что чем меньше размер частицы, тем больше ее удельная поверхность. В промышленности получают высокодисперсные порошки с удельной поверхностью порядка сотен квадратных метров на 1 г вещества, однако из-за их легкой вспыливаемости их чаще используют в качестве наполнителей полимеров, лаков и смазок.
Наиболее распространенные непористые адсорбенты: оксиды  графитированная сажа, белая сажа, аэросил. графитированная сажа, белая сажа, аэросил.
Пористые тела - это твердые тела, внутри которых имеются поры, обусловливающие наличие внутренней межфазной поверхности.
Адсорбция на пористых телах, так же как на непористых, требует достаточного сродства между адсорбентом и адсорбтивом. Однако кроме этого она зависит:
от размеров пор;
от пористости.
В зависимости от размеров пор пористые адсорбенты подразделяют на
а) макропористые, 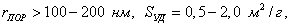 поры играют роль транспортных каналов; поры играют роль транспортных каналов;
б) переходнопористые (капиллярно-пористые), 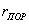 в пределах 1,5-100 нм, в пределах 10-500 в пределах 1,5-100 нм, в пределах 10-500 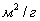 . На стенках этих пор при малых давлениях происходит полимолекулярная адсорбция паров, которая с увеличением давления заканчивается капиллярной конденсацией. Из промышленных адсорбентов к ним относятся силикагели, алюмогели, алюмосиликагели. . На стенках этих пор при малых давлениях происходит полимолекулярная адсорбция паров, которая с увеличением давления заканчивается капиллярной конденсацией. Из промышленных адсорбентов к ним относятся силикагели, алюмогели, алюмосиликагели.
в) микропористые (размеры пор соизмеримы с размерами адсорбированных молекул), в пределах 0,5-1,5 нм, в пределах 500-1000. Противоположные стенки пор так близко расположены друг к другу, что их силовые поля перекрываются и адсорбция происходит во всем объеме пор. К ним относятся цеолиты и активные угли.
Пористость (П) - это отношение объема пор 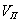 к общему объему тела к общему объему тела 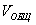 : :
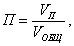
т.е. пористость определяет объем пор, приходящихся на единицу объема тела, т.е. долю пустот в его структуре.
Пористость может измеряться в долях или процентах. Пористость определяет удельную поверхность адсорбента - чем она больше, тем больше емкость адсорбента.
Структура пористого тела в значительной мере влияет на кинетику адсорбции, так как появляется стадия переноса вещества внутри пор. Часто эта стадия определяет время установления равновесия. К числу наиболее распространенных пористых адсорбентов относятся активные угли (их получают из каменного угля, торфа дерева, животных костей, ореховых косточек и др., причем лучшими считаются угли, полученные из скорлупы кокосовых орехов или абрикосовых косточек), силикагели и алюмогели (гидратированные 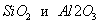 ), цеолиты. ), цеолиты.
Цеолиты (от греч. цео - кипеть, литос - камень) получили свое название за способность эффективно поглощать воду, которая при нагревании испаряется, производя впечатление кипящего камня. Цеолиты являются молекулярными ситами, адсорбирующими лишь определенные компоненты из газовых смесей. Цеолиты представляют собой природные и синтетические алюмосиликаты, имеющие трубчатые полости строго определенного для каждого класса диаметра в диапазоне 0,4-1,1 нм.
Адсорбция газов на твердых поверхностях используется в некоторых отраслях пищевой промышленности, а именно масложировой (например, в производстве маргарина) и в бродильной (например, в производстве дрожжей) для очистки технологических газовых потоков с целью предотвращения выбросов вредных веществ в атмосферу. Поглощение паров воды происходит на пористых веществах, которые выполняют роль твердого адсорбента. Подобные процессы наблюдаются в отношении сахара, соли и сухарей. Адсорбционный способ регулирования газового состава хранилищ скоропортящихся продуктов позволяет в несколько раз сократить потери и увеличить сроки хранения. Адсорбция различных пищевых кислот, лимонной в частности, снижает по сравнению с водой поверхностное натяжение большинства прохладительных напитков. Адсорбция веществ на поверхности раздела жидкость - газ способствует устойчивости пен. Подобный процесс имеет место в бродильной промышленности при производстве дрожжей и некоторых других полупродуктов. Усиление смачивания водой различных поверхностей широко используется в промышленности в качестве сопутствующего процесса при мойке оборудования, подготовке сырья, обработке полуфабрикатов и т.д. Адсорбция на границе твердое тело - жидкость широко применяется при очистке жидкостей (например, диффузионного сока при производстве сахара, растительных масел и соков) от примесей.
| Вопрос |
ответ |
| Какие вещества энергично адсорбируют силикагель? |
1. воду;
2. бензол;
3. спирт;
4. все вышеназванные в 1-3 ответах.
|
Силикаге́ль представляет собой высушенный гель, образующийся из пересыщенных растворов кремниевых кислот (nSiO2mH2O) при pH >5-6. Твёрдый гидрофильный сорбент.
Получается при подкислении растворов силикатов щелочных металлов с последующей промывкой высушиванием образовавшегося геля.
Na2
SiO3
+ 2HCl = 2NaCl + H2
SiO3
H2
SiO3
= SiO2
+ H2
O
Силикагель имеет огромную площадь поверхности (800 м²/1 г), состоящую из групп - SiOH, расположенных на расстоянии 0,5 нм друг от друга. Эти группы являются активными центрами, причём активность конкретной партии силикагеля зависит от числа и активности таких центров. В активном адсорбенте, то есть таком, из которого удалена адсорбированная на его поверхности вода, многие центры будут активны.
Поверхность силикагеля состоит из полярных частиц, то есть она гидрофильна и поэтому энергично адсорбирует воду и слабо адсорбирует неполярные жидкости (углеводороды, эфиры, масла и т.д.). Силикагель хорошо адсорбирует пары полярных веществ, например: спиртов и бензола.
Ответ: п.4
Пены - ячеистые дисперсные системы, представляющие собой совокупность пузырьков газа (пара), разделённых тонкими прослойками жидкости. Пены по размеру пузырьков относятся к грубодисперсным системам; размер пузырьков, составляющих дисперсную фазу, лежит в пределах от долей мм до нескольких см. Общий объём заключённого в них газа может в сотни раз превосходить объём дисперсионной среды - жидкости, находящейся в прослойках.
Образование пены, или вспенивание, происходит при диспергировании газа в жидкой среде и во время выделения новой газовой фазы в объёме жидкости. Возникновение устойчивых высокодисперсных пены обусловлено присутствием в жидкости стабилизаторов пены, или пенообразователей. Эти вещества облегчают вспенивание и затрудняют отток жидкости (дренаж) из пенных плёнок, препятствуя коалесценции (слиянию) пузырьков. Действуют они так же, как стабилизаторы эмульсий и лиофобных коллоидных систем: снижают поверхностное натяжение и создают адсорбционно-сольватный слой с положительным расклинивающим давлением. В водных средах особенно эффективны мыла, мылоподобные поверхностно-активные вещества и некоторые растворимые полимеры, образующие на границе жидкости с газом слои с явно выраженными структурно-механическими свойствами. Увеличение вязкости дисперсионной среды повышает устойчивость пены. Чистые жидкости с низкой вязкостью не образуют пены
Пены по своей природе близки к концентрированным эмульсиям, но дисперсной фазой в них является газ, а не жидкость. Пены получают из растворов поверхностно-активных веществ. Для повышения их устойчивости в растворы ПАВ добавляют высокомолекулярные вещества, повышающие вязкость растворов.
В качестве характеристик пены используется комплекс свойств, всесторонне характеризующих пену.
1) Пенообразующая способность раствора - количество пены, выражаемое её объемом (см3) или высотой столба (м), которое образуется из заданного постоянного объема пенообразующего раствора при соблюдении некоторых стандартных условий пенообразования в течение постоянного времени.
2) Кратность пены b, которая представляет собой отношение объема пены Vп к объему раствора Vж, пошедшего на ее образование (рис.2):
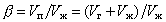 , (2.7.24) , (2.7.24)
где Vг - объем газа в пене.
3) Стабильность (устойчивость) пены - ее способность сохранять общий объем, дисперсность и препятствовать вытеканию жидкости (синерезису). Часто в качестве меры стабильности используют время существования ("жизни") выделенного элемента пены (отдельного пузырька или пленки) или определенного объема пены.
4) Дисперсность пены, которая может быть охарактеризована средним размером пузырьков, распределением их по размерам или поверхностью раздела "раствор - газ" в единице объема пены.
Газовые пузырьки в пенах разделены тончайшими пленками, образующими в своей совокупности пленочный каркас, который и служит основой пен. Такой пленочный каркас образуется, если объем газа составляет 80-90% общего объема. Пузырьки плотно прилегают друг к другу и их разделяет только тонкая пленка раствора пенообразователя. Пузырьки деформируются и приобретают форму пентаэдров. Обычно пузырьки располагаются в объеме пены таким образом, что три пленки между ними соединяются как это показано на рисунке.
В каждом ребре многранника сходятся три пленки, углы между которыми равны 120о. Места стыка пленок (ребра многогранника) характеризуются утолщениями, образующими в поперечном сечении треугольник. Эти утолщения называют каналами Плато-Гиббса, в честь известных ученых - бельгийского ученого Ж. Плато и американского - Дж. Гиббса, внесших большой вклад в изучение пен. Четыре канала Плато-Гиббса сходятся в одной точке, образуя по всей пене одинаковые углы 109о 28’. Площадь поперечного сечения треугольного канала Плато-Гиббса определяется как
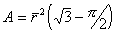 , (2.7.24) , (2.7.24)
где 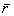 - средний радиус пузырьков в пене. - средний радиус пузырьков в пене.
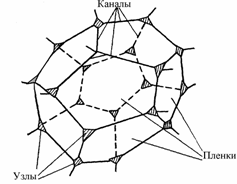
Рисунок 2. Схема фрагмента высокократной пены
Если объем газовой фазы невелик и пленки между пузырьками толстые, то такая пена неустойчива и очень быстро разрушается. В зависимости от формы пузырьков пены делятся на сферические и многогранные. Сферические пены отличаются высоким содержанием жидкости и поэтому неустойчивы, их относят к метастабильным. В таких системах пузырьки коалесцируют - сливаются при соприкосновении.
Пены также весьма распространены среди пищевых продуктов. Чтобы пена образовалась и могла существовать, необходимо присутствие в системе поверхностно-активных веществ - пенообразователей. Эти же вещества чаще всего выполняют и роль стабилизаторов пен. Как и другие коллоидные системы, пены термодинамически нестабильны. Газ и жидкость, из которых они состоят, стремятся образовать два слоя с минимальной поверхностью раздела фаз. Поэтому пены в готовых пищевых продуктах стабилизируют формированием мельчайших кристаллов сахара (нуга), фиксируют путём термообработки (подсушивание зефира, выпекание бисквита, закаливание мороженого) и добавкой стабилизаторов пены.
Пена всегда рассматривалась в кулинарии как нежелательный, побочный элемент, и потому существовали стойкие рекомендации - снимать ее сразу же после появления в любом блюде. Эти рекомендации, обосновывавшиеся прежде, особенно в XVIII-XIX вв., лишь соображениями красоты и аромата кушаний, приобретают сейчас особое гигиеническое и профилактическое значение. К тому же блюда, с которых в процессе приготовления регулярно снимают пену, значительно лучше по вкусу.
Пену, появляющуюся на поверхности воды при приготовлении таких блюд, как супы, компоты, отварные изделия из теста и картофеля, нужно снимать сразу же в момент ее появления и до полного исчезновения. Только после этого суп или компот можно наполнять другими компонентами - пряностями, жирами или сахаром. При появлении пены на поверхности супов следует сразу же уменьшить огонь, чтобы она спокойно сбивалась к краям посуды, а не дробилась и не вваривалась в жидкость.
При изготовлении варенья пену снимают сначала с сахарного сиропа, а затем вторично, после закладки сырья. Чем лучше очищен сироп, тем меньше пены появляется при варке ягод или фруктов. Варенье, с которого быстро снимают пену, лучше и равномернее проваривается и, главное, не переваривается, а потому сохраняет естественный цвет, аромат и вкус.
Если сироп предварительно очищен, то пена, образуемая затем при варке ягод и других компонентов, выглядит более чистой и по ней легче следить за процессом приготовления. Пока пена розоватая или лимонно-желтая, варенье может еще стоять на огне. Как только она начинает темнеть или жухнуть - это сигнал, что варенье переварилось.
В варенье пена снимается не сразу по ее появлении, а лишь тогда, когда обнаружатся участки ее уплотнения с краев посуды или в центре и эта более густая и плотная масса легко отделяется ложкой от поверхности кипящего состава. С этого момента пену следует снимать непрерывно, не давая ей выходить за края посуды. "Убегание" пены отрицательно отражается на качестве варенья: оно лишается пенообразного сиропа - основной среды для варки фруктов, ягод.
1. Ахметов Б.В. Задачи и упражнения по физической и коллоидной химии. - Л.: Химия, 1988. - 240 с.
2. Балязин С.А. Практикум по физической и коллоидной химии: учебное пособие. - М.: Просвещение, 1980. - 271 с.
3. Воюцкий С.С. Курс коллоидной химии: Химия, 1974. - 493 с.
4. Глинка Н.Л. Задачи и упражнения по общей химии: Учебное пособие для вузов/ Под ред.В.А. Рабиновича и Х.М. Рубинной. - М.: Интеграл-Пресс, 2002. - 240с.
5. Грег С., Синг К. Адсорбция, удельная поверхность, пористость. М.: Мир, 1984.310 с.
6. Евстратова К.И., Кижин А.А., Молохова Е.Г. Физическая и коллоидная химия. - М.: Высшая школа, 1990. - 487с.
7. Зимон А.Д. Занимательная коллоидная химия - 3-е изд. - М.: Агар, 2002. - 167с.
8. Зимон А.Д., Лещинко А.Ф. Коллоидная химия. - М.: Агар, 2001. - 317с
9. Киселев А.В. Межмолекулярные взаимодействия в адсорбции и хроматографии. М.: Высш. шк., 1986. - 360 с.
10. Скурихин И.М. Все о пище с точки зрения химика. М Высшая школа, 1991. - 219 с.
11. Фролов Ю.Г. Курс коллоидной химии. Поверхностные явления и дисперсные системы. - М.: Химия, 1989. - 463с.
|
