Санкт-Петербургский государственный технический университет
Методы определения вторичной структуры белков
Пособие для проведения лабораторных работ
на кафедре биофизики физико-механического факультета СПбГТУ
Инфракрасная спектроскопия и спектроскопия кругового дихроизма.
Захаров В.В.
1999
Содержание
Введение
1. Спектры кругового дихроизма белков
1.1 Явление кругового дихроизма
1.2 Методы анализа спектров кругового дихроизма белков
1.3 Работа с пакетом программ STRUCTURE по анализу спектров КД белков
2. Инфракрасные спектры поглощения белков
2.1 Поглощение белков в ИК-области
2.2 Методы анализа ИК-спектров белков
2.3 Работа с пакетом программ STRUC по анализу ИК-спектров белков
Список литературы
Хромофоры белковых молекул (то есть химические группы в молекуле белка, ответственные за поглощение света на определенных длинах волн) можно разделить на три класса: пептидные группы, боковые группы аминокислотных остатков и простетические группы. Спектроскопические методы исследования вторичной структуры белка основаны на изучении спектров именно пептидных хромофоров, поскольку конформация пептидных групп и определяет тот или иной тип вторичной структуры белка - a-спираль, b-структуру и др. Изучение поглощения света пептидными группами белка обычно проводится в ультрафиолетовом и в инфракрасном диапазонах. Как показывают эксперименты, простая адсорбционная спектроскопия белков в неполяризованном ультрафиолетовом свете мало пригодна для анализа вторичной структуры белка. Более ценную информацию можно извлечь из спектров кругового дихроизма белка. Инфракрасные спектры поглощения белка также пригодны для анализа его вторичной структуры [1]. Ниже будет рассмотрено применение методов измерения кругового дихроизма и инфракрасной спектроскопии для анализа вторичной структуры белка.
Белки, как практически все биологические молекулы, вследствие своей пространственной асимметрии обладают оптической активностью. При прохождении через оптически активную среду плоскополяризованный свет становится эллиптически поляризованным. Эллиптичность света q
является одной из мер оптической активности. Она определяется как арктангенс отношения малой и большой осей эллипса. Другим параметром, характеризующим оптическую активность, является отклонение большой оси эллипса от направления поляризации падающего света, называемое оптическим вращением (или дисперсией оптического вращения) j
.
Реклама

Если представить плоскополяризованную волну Е
в виде суммы двух волн противоположной круговой поляризации Е
=Е
L
+Е
R
, то можно показать, что величина j
пропорциональна разности показателей преломления среды для этих волн nL
-nR
, а величина q
- разности коэффициентов экстинции eL
-eR
. Таким образом, оптическое вращение и появление эллиптической поляризации у плоскополяризованного света при прохождении его через оптически активную среду можно объяснить различным замедлением (nL
¹nR
) и поглощением (eL
¹eR
) двух его составляющих Е
L
и Е
R
, поляризованных по кругу. Разность Dn=nL
-nR
называют круговым двулучепреломлением, а разность De=eL
-eR
- круговым дихроизмом. Зависимости этих величин от длины волны называют спектрами дисперсии оптического вращения (ДОВ) и кругового дихроизма (КД).
На самом деле, ДОВ и КД являются проявлениями одного и того же физического явления, а их спектры можно выводить один из другого. Поэтому на практике достаточно измерять лишь один из этих двух спектров. Спектры КД более удобны для использования на практике, поскольку содержат узкие, хорошо разрешимые полосы. Этим объясняется то, что в настоящее время метод измерения КД используется гораздо более широко, чем ДОВ, несмотря на то, что он требует гораздо более сложного экспериментального оборудования.
КД легко измерить путем попеременного пропускания через образец лево - и правополяризованного по кругу света и регистрации соответствующей разницы поглощений, поскольку эллиптичность выходящего из оптически активного образца света обычно очень мала, и ее точное измерение весьма затруднительно. Однако, разность поглощений обычно пересчитывают в значения эллиптичности. Для того, чтобы можно было сравнивать результаты, полученные при исследовании разных образцов, пользуются значениями так называемой молярной эллиптичности:
[q] = 100q
/ Cl
= 3300 De, (1.1.1)
где С
- молярная концентрация, а l
- длина оптического пути.
В случае белков главной целью измерения спектров КД является определение содержания в них вторичных структур разных типов. Если доля ароматических аминокислот в белке не очень велика, его оптическая активность в области от 180 до 240 нм определяется главным образом полипептидным остовом. Многочисленные эксперименты показали, что алифатические боковые группы аминокислотных остатков белка также не дают заметного вклада в спектр КД в этой области. Следовательно, в первом приближении белковую молекулу можно рассматривать просто как комбинацию участков полипептидным остова, находящихся в конформациях a-спирали, b-структуры и беспорядочного клубка.
Реклама

Поглощение света пептидной группой
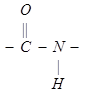
в ультрафиолетовом диапазоне определяется электронными переходами в ее электронных оболочках. В этом процессе основное участие принимают три молекулярных орбитали пептидной группы: n-орбиталь - несвязывающая орбиталь, на которой располагается неподеленная пара 2py
-электронов атома кислорода, p-орбиталь - связывающая орбиталь, на которой располагаются 2pz
-электрон атома кислорода и 2pz
-электрон атома углерода, в значительной степени делокализованные по атомам кислорода, углерода и азота, и p*-орбиталь - разрыхляющая орбиталь, на которой в основном состоянии электроны отсутствуют. Два электронных перехода с наименьшей энергией наблюдаются при возбуждении электрона с n-орбитали на p*-орбиталь (n®p* переход) и с p-орбитали на p*-орбиталь (p®p* переход). n®p* переходу в пептидах соответствует слабая полоса поглощения 210-220 нм, а p®p* переходу гораздо более сильная полоса с максимумом вблизи 190 нм (характерная для a-спиральной конформации).
КД различных типов вторичной структуры белка можно оценить по результатам измерения КД гомополипептидов известной конформации (например, поли-L-лизина), после чего определить вклад каждой из структур в спектр КД исследуемого белка. Однако, такой подход имеет ряд больших недостатков. Во-первых, участки упорядоченной вторичной структуры модельных гомополипептидов имеют значительно большую длину, чем длина типичных участков в глобулярных белках. Во-вторых, их конформация может сильно отличаться от конформации, наблюдаемой у элементов вторичной структуры реальных белков. Кроме этого, среди гомополипептидов нельзя найти "стандартов" для b-изгибов. И, наконец, хотя вклад в КД от взаимодействий между хромофорами уменьшается как квадрат расстояния между ними, должен существовать определенный вклад от взаимодействия между участками с различной вторичной структурой. Эти взаимодействия нельзя адекватно смоделировать, рассматривая протяженные гомополимеры. Поэтому на практике спектры КД гомополипептидов не используются. Вместо этого в качестве базисных берут спектры КД белков, структура которых известна из данных рентгеноструктурного анализа. Различные подходы к анализу исследуемого спектра КД на основе этого базисного набора определяют различия между методами, которые будут описаны ниже.
Метод "эталонных спектров" [2,3]. М
етоды предсказания вторичной структуры белков по их спектрам КД основаны на предположении о том, что спектры КД различных структурных форм, составляющих белковую молекулу, дают аддитивный вклад в спектр КД белка в целом. Это можно записать в следующем виде:
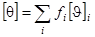 (1.2.1) (1.2.1)
где  - спектр КД белка (зависимость эллиптичности от длины волны света), - спектр КД белка (зависимость эллиптичности от длины волны света), 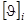 - идеализированный "эталонный спектр" - спектр КД, соответствующий i
-ой структурной форме, участвующей в образовании вторичной структуры белка, - идеализированный "эталонный спектр" - спектр КД, соответствующий i
-ой структурной форме, участвующей в образовании вторичной структуры белка, 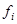 - доля этой формы во вторичной структуре, причем - доля этой формы во вторичной структуре, причем
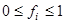 и и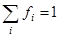 . (1.2.2) . (1.2.2)
Эталонные спектры для всех структурных форм могут быть вычислены на основании набора базисных спектров КД (спектров белков с известной вторичной структурой - коэффициентами ) с помощью метода наименьших квадратов и формулы (1.2.1), примененной к каждому базисному спектру. После этого экспериментальный спектр КД исследуемого белка с помощью того же метода наименьших квадратов может быть аппроксимирован по формуле (1.2.1) с использованием вычисленных эталонных спектров. При этом вклад каждого из эталонных спектров будет равен доле соответствующей ему структурной форме в общей структуре белка Такой подход к анализу спектров КД белков был впервые использован в работе [2]. Ниже будет более подробно рассмотрена модификация этого метода [3].
Принимая в рассмотрение в качестве структурных классов a-спираль (H), b-структуру (b), b-изгиб (t) и “неупорядоченную” форму (R), можем написать:
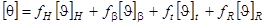 . (1.2.3) . (1.2.3)
Суммируя экспериментальные данные, вместо 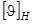 в уравнение (1.2.3) вводят величину в уравнение (1.2.3) вводят величину 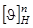 , учитывающую зависимость эталонного спектра, соответствующего a-спирали, от числа аминокислотных остатков, образующих ее: , учитывающую зависимость эталонного спектра, соответствующего a-спирали, от числа аминокислотных остатков, образующих ее:
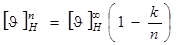 , (1.2.4) , (1.2.4)
где и 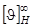 - эталонные спектры для a-спирали из n
аминокислотных остатков и для a-спирали “бесконечной” длины, а k
- так называемый фактор длины цепи ( - эталонные спектры для a-спирали из n
аминокислотных остатков и для a-спирали “бесконечной” длины, а k
- так называемый фактор длины цепи (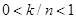 ). Согласно теоретическим расчетам оптической активности a-спирали и экспериментальным данным, спектр КД a-спирали в диапазоне 185-240 нм может быть разложен на три независимых оптически активных составляющих (n®p*, p®p||
*, p®p^
*), которые можно описать гауссовскими зависимостями: ). Согласно теоретическим расчетам оптической активности a-спирали и экспериментальным данным, спектр КД a-спирали в диапазоне 185-240 нм может быть разложен на три независимых оптически активных составляющих (n®p*, p®p||
*, p®p^
*), которые можно описать гауссовскими зависимостями:
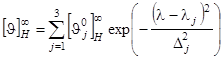 , (1.2.5) , (1.2.5)
где 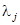 и и 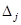 - положение максимума и полуширина j
-ой гауссовской линии в спектре КД a-спирали, а - положение максимума и полуширина j
-ой гауссовской линии в спектре КД a-спирали, а 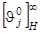 - максимальное значение эллиптичности “бесконечной” a-спирали на длине волны . В окончательном виде для спектра КД белка можно написать следующее выражение: - максимальное значение эллиптичности “бесконечной” a-спирали на длине волны . В окончательном виде для спектра КД белка можно написать следующее выражение:
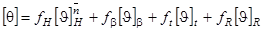 , (1.2.6) , (1.2.6)
где
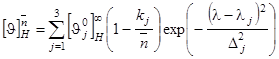 . (1.2.7) . (1.2.7)
Здесь 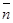 - среднее число аминокислот на a-спиральный участок цепи в молекуле белка. - среднее число аминокислот на a-спиральный участок цепи в молекуле белка.
Параметры , , и 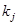 в уравнении (1.2.7) были найдены на основе спектра КД миоглобина. Они имеют следующие значения: в уравнении (1.2.7) были найдены на основе спектра КД миоглобина. Они имеют следующие значения:
| j
|
, нм |
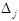 , нм , нм |
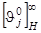 , град×см2
×дмоль-1 , град×см2
×дмоль-1
|
|
| 1 |
223.4 |
10.8 |
-3.73×10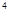 |
2.50 |
| 2 |
206.6 |
8.9 |
-3.72×10 |
3.50 |
| 3 |
193.5 |
8.4 |
+10.1×10 |
2.50 |
Эти параметры для глобулярных белков с достаточно большой точностью можно считать постоянными. Попытки оценить 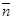 для конкретных белков по их спектрам КД оказались ненадежными. Для большинства исследованных белков этот параметр оказался равным примерно 10-11 аминокислотам на a-спиральный сегмент. Распространяя этот факт на все анализируемые белки, авторы данного метода положили равным 10. для конкретных белков по их спектрам КД оказались ненадежными. Для большинства исследованных белков этот параметр оказался равным примерно 10-11 аминокислотам на a-спиральный сегмент. Распространяя этот факт на все анализируемые белки, авторы данного метода положили равным 10.
Вклад b-структуры в спектр КД белка оказывается зависящим от гораздо большего числа параметров: не только от числа аминокислотных остатков на сегмент, но и от числа нитей в данном участке структуры и их направленности, поэтому его описание простым уравнением, подобным уравнению (1.2.7), невозможно. То же самое касается b-изгиба и, особенно, “неупорядоченной” формы, под которой подразумевается все, не относящееся к другим классам. Используемые в данном методе эталонные спектры b-структуры, b-изгиба и “неупорядоченной” формы являются статистически усредненными по белкам, используемым в качестве базисных.
Процедура анализа спектра КД исследуемого белка подразделяется на два этапа. Первый этап заключается в вычислении эталонных спектров структурных элементов, то есть значений 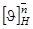 , , 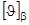 , , 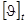 и и 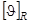 для длин волн в диапазоне 185-240 нм с интервалом в 1 нм, на основе экспериментальных спектров КД пятнадцати эталонных белков со значениями для длин волн в диапазоне 185-240 нм с интервалом в 1 нм, на основе экспериментальных спектров КД пятнадцати эталонных белков со значениями 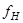 , , 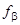 , , 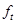 , , 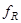 , , известными из рентгеноструктурного анализа. Эталонный спектр, соответствующий a-спирали, может быть вычислен непосредственно по формуле (1.2.7). Остальные эталонные спектры находятся из уравнения (1.2.6) с помощью метода наименьших квадратов, причем для уменьшения числа неизвестных в этом уравнении из экспериментального спектра КД каждого эталонного белка исключается вклад a-спиральной формы, вычисленный по формуле (1.2.7). Эталонные спектры, вычисленные с помощью данного метода показаны на рисунке 1.2.1. , , известными из рентгеноструктурного анализа. Эталонный спектр, соответствующий a-спирали, может быть вычислен непосредственно по формуле (1.2.7). Остальные эталонные спектры находятся из уравнения (1.2.6) с помощью метода наименьших квадратов, причем для уменьшения числа неизвестных в этом уравнении из экспериментального спектра КД каждого эталонного белка исключается вклад a-спиральной формы, вычисленный по формуле (1.2.7). Эталонные спектры, вычисленные с помощью данного метода показаны на рисунке 1.2.1.
Когда эталонные спектры найдены, могут быть вычислены коэффициенты , , , в уравнении (1.2.6), примененном к спектру КД исследуемого белка. Для этого также используется метод наименьших квадратов. Он заключается в подборе таких коэффициентов , что
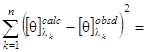 minimum. (1.2.8) minimum. (1.2.8)
Здесь 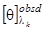 - экспериментальный, а - экспериментальный, а 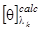 - рассчитанный по формуле (1.2.6) спектр КД исследуемого белка; - рассчитанный по формуле (1.2.6) спектр КД исследуемого белка; 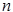 - число точек в спектре. Коэффициенты , являющиеся решением уравнения (1.2.8) с учетом условий (1.2.2), представляют собой искомые доли структурных элементов во вторичной структуре белка. - число точек в спектре. Коэффициенты , являющиеся решением уравнения (1.2.8) с учетом условий (1.2.2), представляют собой искомые доли структурных элементов во вторичной структуре белка.
Метод "регуляризации" [4].
Подход к анализу спектра КД белка, лежащий в основе предыдущего метода, заключается в определении эталонных спектров, которые, как можно было бы предполагать, полностью характеризуют структурные элементы, образующие вторичную структуру исследуемого белка. Однако, как показывают экспериментальные данные, ни один эталонный спектр не может точно описать все разновидности таких обширных и достаточно неопределенных классов, как a-спираль, b-структура, b-изгиб и др.
Конформация элементов вторичной структуры глобулярных белков значительно отличается от идеальной. Кроме этого, вклад каждого структурного класса в спектр КД белка зависит от очень многих параметров, о которых упоминалось выше. Для учета всего разнообразия типов вторичной структуры белков требуется расширить исходный набор базисных спектров. В результате возникающей при этом избыточности начальных данных обычный метод наименьших квадратов становится неустойчивым к экспериментальной ошибке и приводит к заведомо неверным результатам. Применение метода "эталонных спектров" в том виде, как он описан в предыдущем пункте, к большому базисному набору спектров оказывается, по сути, некорректным.
Эту проблему частично можно разрешить, заменив метод наименьших квадратов моделью, применение которой, на первый взгляд, не вполне оправдано и адекватно, но зато приводит к устойчивому к экспериментальной ошибке результату даже в случае большого числа параметров. Применение такой стабилизирующей модели позволяет подойти к анализу спектров КД с другой стороны. А именно, появляется возможность прямого представления спктра КД исследуемого белка в виде линейной комбинации базисных спектров. Таким образом удается полностью избежать проблемы, связанной с определением эталонных спектров отдельных структурных классов и проводить более гибкий и точный анализ с использованием реальных белковых спектров.
Рассмотрим данный метод более подробно. Предположим, что нам удалось представить спектр КД исследуемого белка в виде линейной комбинации спектров 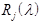 базисных белков, структура которых известна из рентгеноструктурного анализа. Обозначим число этих спектров через базисных белков, структура которых известна из рентгеноструктурного анализа. Обозначим число этих спектров через 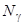 (в данном методе =16). Тогда можем записать: (в данном методе =16). Тогда можем записать:
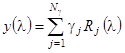 , (1.2.9) , (1.2.9)
где 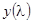 - спектр КД (эллиптичность) исследуемого белка. - спектр КД (эллиптичность) исследуемого белка.
Обозначим долю аминокислот j
-ого базисного белка в i
-ом структурном классе через 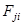 , тогда базисные спектры могут быть представлены в виде суперпозиции , тогда базисные спектры могут быть представлены в виде суперпозиции 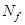 идеализированных эталонных спектров идеализированных эталонных спектров 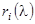 , соответствующих отдельным структурным классам: , соответствующих отдельным структурным классам:
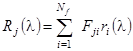 . (1.2.10) . (1.2.10)
Аналогично для спектра КД исследуемого белка:
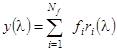 . (1.2.11) . (1.2.11)
Подставляя равенства (1.2.10) и (1.2.11) в уравнение (1.2.9), получим связь искомых коэффициентов 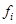 с известными (из рентгеноструктурного анализа) коэффициентами : с известными (из рентгеноструктурного анализа) коэффициентами :
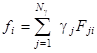 . (1.2.12) . (1.2.12)
Проблема заключается в определении коэффициентов 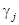 в разложении (1.2.9). В подобных задачах широко применяется метод наименьших квадратов, определяющий коэффициенты из следующего условия: в разложении (1.2.9). В подобных задачах широко применяется метод наименьших квадратов, определяющий коэффициенты из следующего условия:
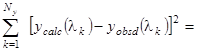 minimum (1.2.13) minimum (1.2.13)
с ограничениями
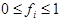 и и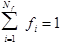 . (1.2.14) . (1.2.14)
Здесь 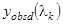 и и 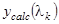 - экспериментальное и рассчитанное по формуле (1.2.9) значения для эллиптичности на длине волны - экспериментальное и рассчитанное по формуле (1.2.9) значения для эллиптичности на длине волны 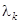 , , 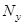 - число точек в спектре. - число точек в спектре.
Согласно теореме Гаусса-Маркова, среди линейных несмещенных оценок оценка, получаемая с помощью метода наименьших квадратов, является наиболее эффективной в том смысле, что рассчитанные с его помощью коэффициенты наиболее близки к своим истинным значениям. Однако, при больших значениях метод наименьших квадратов становится крайне неустойчивым к экспериментальной ошибке. Повышение стабильности метода за счет снижения величины , в свою очередь, также приводит к заметной ошибке.
Авторы метода [4] нашли выход в использовании вместо метода наименьших квадратов линейной смещенной оценки, определяемой следующим условием:
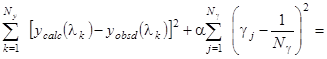 minimum. (1.2.15) minimum. (1.2.15)
Эта оценка является смещенной и, следовательно, приводит к систематической ошибке. Тем не менее при больших значениях она дает значения более близкие реальным, чем получаемые с помощью метода наименьших квадратов. Очевидно, что уравнение (1.2.15) также необходимо дополнить условиями (1.2.14).
Рассмотрим критерий (1.2.15) более подробно. При a=0 мы получаем обычный метод наименьших квадратов, не пригодный в нашем случае. При a>0 второй член в левой части (1.2.15) является регуляризатором. Он стабилизирует решение, поддерживая коэффициенты малыми (близкими к 1/). Тем не менее, если некоторый спектр 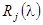 содержит компоненты, которые хорошо аппроксимируют , это ограничение не будет иметь такой силы, так как минимизация левой части уравнения (1.2.15) сможет быть достигнута в большей степени уменьшением первого члена, чем второго, что приводит к наиболее оптимальному значению . Таким образом получается очень гибкая, но стабильная модель, которая самостоятельно выбирает из большого набора базисных спектров те, которые аппроксимируют данные наилучшим образом. В случае анализа спектров КД белков уравнению (1.2.15) можно дать следующую интерпретацию. Поскольку априори нельзя сказать, какой из спектров будет аппроксимировать лучше, ни один из них не имеет преимущества, и все коэффициенты полагаются приблизительно равными, близкими к 1/ (смотри условия (1.2.14)). содержит компоненты, которые хорошо аппроксимируют , это ограничение не будет иметь такой силы, так как минимизация левой части уравнения (1.2.15) сможет быть достигнута в большей степени уменьшением первого члена, чем второго, что приводит к наиболее оптимальному значению . Таким образом получается очень гибкая, но стабильная модель, которая самостоятельно выбирает из большого набора базисных спектров те, которые аппроксимируют данные наилучшим образом. В случае анализа спектров КД белков уравнению (1.2.15) можно дать следующую интерпретацию. Поскольку априори нельзя сказать, какой из спектров будет аппроксимировать лучше, ни один из них не имеет преимущества, и все коэффициенты полагаются приблизительно равными, близкими к 1/ (смотри условия (1.2.14)).
При возрастании параметра a точность аппроксимации экспериментальных данных падает за счет уменьшения эффективного числа степеней свободы, соответствующего числу свободных параметров в обычном методе наименьших квадратов. Обычно при малых a это происходит медленно, но когда этот параметр становится слишком большим, число степеней свободы становится таким малым, что коэффициенты становятся равными 1/, и метод полностью теряет свою гибкость. Выбор параметра a определяется оптимальным компромиссом между гибкостью и стабильностью модели, тем самым давая наилучшие значения . Авторы данного метода осуществляли выбор a с помощью автоматического статистического теста на относительное увеличение отклонения аппроксимирующего спектра (реконструированного из спектров эталонных белков) от экспериментальных данных при увеличении этого параметра.
Если при анализе спектра КД белка нам известно, что среди белков базисного набора есть белки, структурно схожие с исследуемым, то в уравнение (1.2.15) можно ввести эти данные с помощью различного взвешивания отдельных членов второй суммы этого уравнения, тем самым давая соответствующим коэффициентам большую свободу изменения. Однако сделать это объективно и количественно довольно сложно, поэтому авторы метода не пользовались этим. Как показывают эксперименты, в случае структурной схожести белков соответствующие коэффициенты автоматически выбираются наибольшими без какой-либо дополнительной информации.
Метод "ортогональных спектров" [5,6]. О
сновой данного метода является метод собственных векторов многокомпoнентного матричного анализа. Он позволяет проводить быструю обработку больших наборов данных с помощью формирования из них ортогональных компонент в виде собственных векторов с соответствующими собственными значениями.
Этот метод использует в качестве базисных спектры КД 16 белков с известной вторичной структурой в диапазоне 178-260 нм с интервалом в 2 нм (всего по 42 точки в каждом из 16 спектров). Пусть С
- прямоугольная матрица размером 16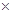 42, содержащая в качестве строк спектры КД эталонных белков. Умножая ее на свою транспонированную матрицу, получим симметричную квадратную матрицу CC
T
размером 1616. Приведем эту матрицу к диагональному виду с помощью ортогональной матрицы U
(1616): 42, содержащая в качестве строк спектры КД эталонных белков. Умножая ее на свою транспонированную матрицу, получим симметричную квадратную матрицу CC
T
размером 1616. Приведем эту матрицу к диагональному виду с помощью ортогональной матрицы U
(1616):
(CC
T
) U = UE
. (1.2.16)
Матрица U
будет состоять из 16 собственных векторов, а диагональная матрица Е -
из 16 собственных значений матрицы CC
T
. Рассмотрим матрицу B
, определяемую выражением
B = U
T
C
. (1.2.17)
Это прямоугольная матрица, которая, также как и матрица исходных спектров КД базисных белков, имеет размер 1642. Ее строки можно использовать в качестве 16 новых ортогональных базисных спектров КД, каждый из которых представляет собой линейную комбинацию исходных белковых спектров. Разложение произвольного спектра КД по этим новым базисным спектрам, вместо исходных, оказывается более удобным, поскольку “значимость" каждого их них в этом разложении, то есть степень, в которой он представляет исходный набор базисных спектров, пропорциональна квадратному корню из соответствующего собственного значения. Из этого следует, что любой неполный набор из ортогональных базисных спектров, выбранный таким образом, что соответствующие им собственные значения максимальны, будет описывать произвольный белковый спектр КД лучше, чем любой неполный набор из исходных спектров базисных белков.
Ошибка, возникающая при аппроксимации экспериментального белкового спектра КД с помощью неполного набора наиболее “значимых" ортогональных базисных спектров, определяется следующей формулой:
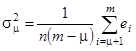 . (1.2.18) . (1.2.18)
Здесь s - среднее квадратичное отклонение, n - число точек в спектре, m - число базисных спектров в исходном наборе, 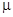 - число ортогональных базисных спектров в неполном наборе, используемом для реконструкции произвольного белкового спектра, а - число ортогональных базисных спектров в неполном наборе, используемом для реконструкции произвольного белкового спектра, а 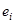 - собственные значения, расположенные в ряд в порядке убывания их величины. Случайная ошибка, связанная с погрешностью измерений при снятии спектров КД эталонных белков, приблизительно равна 0.3 единицы De. Сравним ее со значениями s, рассчитанными по формуле (1.2.18) для разных значений m (при m=16): - собственные значения, расположенные в ряд в порядке убывания их величины. Случайная ошибка, связанная с погрешностью измерений при снятии спектров КД эталонных белков, приблизительно равна 0.3 единицы De. Сравним ее со значениями s, рассчитанными по формуле (1.2.18) для разных значений m (при m=16):
| m |
s, ед. De |
| 3 |
0.38 |
| 4 |
0.24 |
| 5 |
0.17 |
| 6 |
0.12 |
Из приведенной таблицы видно, что четыре ортогональных базисных спектра дают значение s, нe превышающее уровень случайной ошибки. Но эксперименты показывают, что форма реконструированного таким образом спектра плохо совпадает с реальной. Пять ортогональных базисных спектров дают значение s, в два раза меньшее уровня случайной ошибки, и при этом хорошо воспроизводят форму спектра. Шесть ортогональных базисных спектров дают лишь незначительное улучшение.
Это объясняется тем, что оставшиеся базисные спектры представляют собой ни что иное, как “шум”, и их учет приводит лишь к увеличению ошибки при вычислениях. Авторы данного метода использовали для вычислений пять "наиболее значимых" ортогональных базисных спектров (m=5), полагая это количество оптимальным. Эти спектры представлены на рисунке 1.2.2.
Из выражения (1.2.17) следует, что
С = UB
. (1.2.19)
Восстанавливая по сокращенному набору ортогональных базисных спектров исходный набор базисных спектров КД, можем написать:
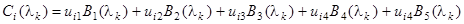 , (1.2.20) , (1.2.20)
где 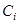 - исходные базисные спектры (i
=1,., 16; k
=1,.,42), а - исходные базисные спектры (i
=1,., 16; k
=1,.,42), а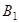 - -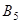 - пять "наиболее значимых" ортогональных базисных спектров. Эксперименты по воспроизведению исходных белковых спектров по формуле (1.2.20) показывают, что среднеквадратичная ошибка при этом составляет от 0.08 до 0.25, что является весьма хорошим показателем. - пять "наиболее значимых" ортогональных базисных спектров. Эксперименты по воспроизведению исходных белковых спектров по формуле (1.2.20) показывают, что среднеквадратичная ошибка при этом составляет от 0.08 до 0.25, что является весьма хорошим показателем.
Представим данные рентгеноструктурного анализа для 16 базисных белков в виде матрицы S
размером 168, содержащей величины относительного содержания в каждом из белков восьми структурных элементов: спиральной структуры, включая a - и 310
-спирали, антипараллельной и параллельной b-структуры, b-изгибов I, II, III типов, других видов b-изгибов и оставшейся (“неупорядоченной”) структуры.
Как можно предполагать из того факта, что исходный набор базисных спектров может быть полностью восстановлен но основе лишь пяти спектров ортогонального базисного набора, спектры КД белков в диапазоне от 178 до 260 нм содержат в себе информацию лишь о пяти независимых типах вторичной структуры.
С точки зрения независимости спектров КД в качестве таких типов вторичной структуры могут быть приняты комбинации обычных типов вторичной структуры (a-спирали, b-структуры и т.д.), соответствующие пяти "наиболее значимым" ортогональным базисным спектрам.
Если для ортогональных базисных спектров также ввести матрицу структурных данных D
(168), то аналогично формуле (1.2.19) можно записать
S = UD
(1.2.21)
Как показывает эксперимент, структурная матрица S
может быть полностью восстановлена на основе лишь пяти комбинаций элементов вторичной структуры матрицы D
, соответствующих пяти "наиболее значимым" ортогональным базисным спектрам. Таким образом, эти комбинации обычных типов вторичной структуры являются (с точки зрения независимости спектров КД) независимыми вторичными "суперструктурами":
| Номер "супер-структуры" |
a, 310
|
b
¯
|
b
|
b-изг.
I
|
b-изг.
II
|
b-изг.
III
|
b-изг.
др.
|
Ост.
типы
|
| 1 |
1.77 |
0.30 |
0.20 |
0.16 |
0.07 |
0.12 |
0.14 |
1.06 |
| 2 |
0.56 |
-0.47 |
-0.06 |
-0.04 |
-0.07 |
-0.01 |
-0.09 |
-0.76 |
| 3 |
0.06 |
0.38 |
-0.12 |
0.01 |
0.02 |
0.01 |
0.01 |
-0.18 |
| 4 |
0.00 |
0.06 |
0.27 |
-0.04 |
-0.02 |
0.00 |
0.03 |
-0.06 |
| 5 |
-0.01 |
-0.01 |
0.02 |
0.16 |
0.02 |
0.05 |
0.00 |
-0.03 |
Следовательно, восемь рассматриваемых в данном методе стандартных структурных классов, вообще говоря, не являются строго независимыми, так как все они также могут быть описаны с помощью пяти независимых “суперструктур”, описанных выше.
Для применения данного метода к анализу спектров КД произвольных белков необходимо, чтобы анализируемый спектр также быть снят в диапазоне от 178 до 260 нм. Поскольку при его аппроксимации базисными спектрами рассматривается лишь небольшой их набор, то проблемы, связанной с неустойчивостью метода наименьших квадратов, не возникает. Однако, очевидно, что приемлемые результаты возможно получить только в том случае, если структурные характеристики исследуемого белка достаточно хорошо представлены среди базисных белков. Для установления достоверности полученных результатов авторы метода рекомендуют использовать метод наименьших квадратов без ограничений на коэффициенты разложения (смотри условия (1.2.2)). При этом большие по модулю отрицательные коэффициенты 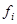 или большое отклонение их суммы от единицы свидетельствуют о том, что метод в данном случае неприменим. Подробнее об этом критерии будет говориться в следующем разделе. или большое отклонение их суммы от единицы свидетельствуют о том, что метод в данном случае неприменим. Подробнее об этом критерии будет говориться в следующем разделе.
Метод "выбора переменных" [7]. О
бычный метод наименьших квадратов, используемый для представления произвольного спектра КД в виде линейной комбинации базисных спектров, имеет по сравнению с другими методами наибольшую гибкость. Это проявляется в том, что спектры базисных белков участвуют в разложении в различной степени в зависимости от характера конкретного спектра. Однако, эксперименты показывают, что наилучшее воспроизведение формы спектра не всегда дает лучшие результаты. Более того, метод наименьших квадратов оказывается неустойчивым к экспериментальной ошибке, если число используемых в разложении базисных спектров превышает информационное содержание анализируемого спектра (для спектров в диапазоне 178-260 нм оно приблизительно равно пяти, а в диапазоне 190-260 нм - четырем).
Метод "регуляризации" [4] решает эту проблему с помощью "регуляризатора", который стабилизирует систему, оставляя ей при этом значительную гибкость. Метод "ортогональных спектров" [5,6] достигает устойчивости метода наименьших квадратов за счет использования только пяти ортогональных базисных спектров, построенных на основе исходного набора спектров базисных белков. Однако, поскольку базисные спектры построены на основе фиксированного набора спектров базисных белков, степень участия последних при воспроизведении анализируемого спектра также оказывается в некоторой мере фиксированной, а гибкость метода - крайне низкой.
Метод "выбора переменных", суть которого будет описана ниже, основан на методе "ортогональных спектров", но обладает значительной гибкостью, достигаемой за счет использования при построении ортогональных базисных спектров различных наборов базисных белков, выбираемых с помощью статистической процедуры "выбора переменных". Рассмотрим смысл этой процедуры более подробно.
Предсказание вторичной структуры белка по его спектру КД должно удовлетворять двум важным условиям:
1. Величины содержания в белке рассматриваемых структурных элементов не должны быть отрицательными: 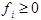 . .
2. Суммарное содержание в белке всех рассматриваемых типов структур должно быть равно единице (100%): 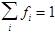 . .
Второе условие является особенно важным при анализе конформационных изменений белка при денатурации или связывании каких-либо лигандов. Во всех методах, описанных выше, оба эти условия вводятся непосредственно в процедуру нахождения коэффициентов  с помощью метода наименьших квадратов. Однако такое ограничение на коэффициенты может весьма заметным образом исказить результаты этой процедуры. с помощью метода наименьших квадратов. Однако такое ограничение на коэффициенты может весьма заметным образом исказить результаты этой процедуры.
Для преодоления подобных недостатков авторы рассматриваемого метода не пользуются условиями (1) и (2) и допускают существование отрицательных коэффициентов и отклонение их суммы от единицы. Появление подобных несоответствий свидетельствует о неуспехе метода и может быть объяснено наличием у некоторых базисных белков таких структурных форм, вкладов которых в спектр исследуемого белка не было обнаружено. Для избежания подобных ситуаций вводится процедура "выбора переменных", которая поочередно исключает белки из исходного базисного набора, а затем проводит вычисления с каждой из полученных комбинаций базисных белков, используя метод "ортогональных спектров". Эксперименты показали, что достоверность результатов значительно повышается по мере того, как сумма коэффициентов приближается к единице. Повышение точности анализа было достигнуто даже при анализе спектров в укороченном диапазоне (190-260 нм).
Поскольку заранее не известно, какие из базисных белков содержат элементы, отсутствующие у исследуемого белка, и спектры которых необходимо исключить из исходного набора для улучшения результатов, рассматриваются все возможные комбинации из исходного набора 16 базисных спектров. Эта процедура выполняется в следующем порядке. Сначала из исходного набора исключаются поочередно по три базисных спектра на каждом шаге, а ортогональные базисные спектры строятся на основе оставшихся 13 исходных базисных спектров. Сравнение результатов, полученных для различных наборов из 13 базисных белков, выявляет один или два белка, которые являлись причиной отклонений коэффициентов и их суммы от условий (1) и (2). Эти белки исключаются из исходного набора, и процедура повторяется до тех пор, пока не будут получены удовлетворительные результаты.
Критериями удовлетворительного решения, соответствующего оптимальному набору базисных спектров, являются следующие условия:
1. Сумма коэффициентов должна находиться в диапазоне от 0.96 до 1.05 (или, по крайней мере, от 0.90 до 1.10).
2. Значение содержания произвольной структурной формы в исследуемом белке () должно быть выше - 0,05.
3. Воспроизведение анализируемого спектра на основе выбранного набора базисных спектров должно быть лучше, чем при использовании полного их набора.
4. Более предпочтительным является набор, содержащий большее число базисных спектров.
5. Более предпочтительными являются те белки, спектры которых ближе к анализируемому спектру.
На практике в большинстве случаев удовлетворительных результатов удается достичь при исключении из исходного набора всего трех или четырех белков, причем среднеквадратичная ошибка при воспроизведении анализируемого спектра составляет меньше 0.2 единицы De. Если несколько наборов базисных белков оказываются удовлетворительными в одинаковой степени, то результаты, полученные на их основе, усредняются.
В заключение можно отметить, что метод "выбора переменных" является мощным средством анализа спектров КД белков в ситуациях, когда другие распространеннные методы дают заведомо неверные результаты.
Сравнение различных методов анализа спектров КД.
Поскольку все методы анализа спектров КД имеют чисто эмпирический характер, каждый из них нуждается в экспериментальной проверке на белках с известными рентгеноструктурными данными. Обычно подобная проверка проводится на белках, включенных в базисный набор для данного метода. При этом белки поочередно исключаются по одному из этого набора, а их спектры анализируются на основе спектров оставшихся белков. После этого результаты, полученные для каждого типа вторичной структуры, сравниваются со значениями, полученными при рентгеноструктурном анализе, с помощью подсчета коэффициента корреляции между этими двумя наборами данных, определяемого следующим выражением:
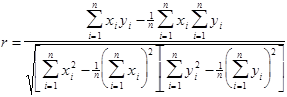 .(1.2.22) .(1.2.22)
Здесь 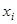 и и 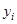 - экспериментальный и рассчитанный наборы данных, n - число белков в базисном наборе. Значения коэффициента корреляции r лежат в диапазоне от - 1 до 1, причем значеия r, близкие к 1, свидетельствуют об успешном предсказании, характеризующимся достаточно высокой точностью. Значения r, близкие к 0 или - 1, говорят о случайном совпадении или полном несоответствии рассчитанных и экспериментальных данных. - экспериментальный и рассчитанный наборы данных, n - число белков в базисном наборе. Значения коэффициента корреляции r лежат в диапазоне от - 1 до 1, причем значеия r, близкие к 1, свидетельствуют об успешном предсказании, характеризующимся достаточно высокой точностью. Значения r, близкие к 0 или - 1, говорят о случайном совпадении или полном несоответствии рассчитанных и экспериментальных данных.
Ниже приведены значения коэффициентов корреляции для четырех рассмотренных методов: метода "эталонных спектров" [2,3], метода "регуляризации" [4], метода "ортогональных спектров" [5,6] и метода "выбора переменных" [7]:
| метод |
диапазон, |
коэффициент корреляции r |
| нм |
a |
b¯ |
b |
b¯+ |
b-изг. |
Ост. |
| [2,3] |
190-240 |
0.85 |
- |
- |
0.25 |
-0.31 |
0.46 |
| [4] |
190-240 |
0.96 |
- |
- |
0.94 |
0.31 |
0.49 |
| [5,6] |
190-260 |
0.98 |
0.40 |
0.00 |
-0.27 |
0.18 |
0.24 |
| [7] |
190-260 |
0.95 |
0.57 |
0.47 |
0.45 |
0.54 |
0.69 |
| [4] |
178-260 |
0.96 |
0.23 |
0.39 |
0.12 |
0.51 |
0.64 |
| [5,6] |
178-260 |
0.98 |
0.55 |
0.63 |
0.54 |
0.30 |
0.61 |
| [7] |
178-260 |
0.97 |
0.78 |
0.67 |
0.76 |
0.49 |
0.86 |
Пакет программ STRUCTUREразработан в институте белка РАН (1991-1992 К.С. Василенко). Он предназначен для анализа спектров кругового дихроизма белков и определения их вторичной структуры. Алгоритм анализа спектров основан на методах, описанных выше. Пакет STRUCTUREсостоит из следующих программ и вспомогательных файлов:
- STRUCTURE (файл structur.exe) - программа, обеспечивающая интерфейс для всех программ пакета, позволяющая также создавать и редактировать файлы данных в универсальном для всех программ формате.
- CONTIN (файл contin.exe) - программа, определяющая вторичную структуру белка методом "регуляризации" [4].
- PROVCD (файл provcd.exe) - программа, осуществляющая проведение статистического теста для программы CONTIN.
- DEF_CLASS (файл def_clas.exe) - программа, определяющая тип третичной структуры белка.
- CDESTIMATE (файл cdestima.exe) - программа, определяющая вторичную структуру белка методом "эталонных спектров" [3].
- VARSELEC (файл varselec.exe) - программа, определяющая вторичную структуру белка методом "ортогональных спектров" с процедурой "выбора переменных" [7].
- RUN.BAT - командный файл, используемый для запуска программ пакета в условиях недостаточного объема оперативной памяти.
- *.DAT - файл, содержащий спектр КД белка, а также данные о его вторичной структуре (если они известны).
- *.GRP - файл, содержащий список базисных спектров КД (принадлежащих одной из базисных групп).
- *.STR - файл, содержащий набор структурных типов (элементов вторичной структуры белка).
После запуска файла structur.exeна экране появляется главное меню программы, состоящее из следующих пунктов:
1. File - создание и редактирование файлов данных;
2. Group - создание и редактирование групп базисных спектров КД белков;
3. Calculate - выбор метода анализа, анализируемого спектра, группы базисных спектров, запуск вычислений и просмотр результатов;
4. Options - выбор набора структурных типов;
5. Setup - изменение цветового оформления окон программы;
6. Quit - выход из программы.
В нижней части экрана располагаются три окна, содержащие информацию об анализируемом спектре КД (Protein), а также о выбранных для анализа группе базисных спектров (Group) и наборе типов вторичной структуры белка (Structures).
Создание и редактирование файлов данных
. Создание и редактирование файлов данных осуществляется с помощью команд меню File/Createи File/Editсоответственно. В файл необходимо внести следующую информацию:
- Комментарий длиной не более 45 символов (пункт меню Comment).
- Идентификатор длиной не более 7 символов, который становится именем файла и автоматически приобретает расширение.dat (пункт меню Identificator).
- Содержание в белке (относительные доли) различных типов вторичной структуры по данным рентгеноструктурного анализа (пункт меню Structuredata). Эти данные необходимы только в случае использования вводимого спектра в дальнейшем в качестве базисного.
- Диапазон и шаг по длинам волн, а также сам спектр КД (пункт меню Spectrum). Для программы CDESTIMATEдиапазон анализируемого спектра не должен быть шире, чем 240 - 190 нм, а шаг должен быть равен 1 нм или больше. Для программы CONTINчисло точек в анализируемом спектре не должно превышать 51. Для программ CONTIN, VARSELECи PROVCDдиапазон анализируемого спектра не должен быть шире диапазона базисных спектров, а шаг должен совпадать с шагом базисных спектров.
После ввода всей перечисленной выше информации необходимо сохранить ее с помощью пункта меню Save. При необходимости можно построить введенный спектр КД на экране в графическом виде с помощью пункта меню View.
Команды меню File/Loadи File/Deleteиспользуются соответственно для добавления новых спектров в список рабочих спектров, запоминаемых программой, и для удаления из него ненужных спектров. Для добавления нового спектра с помощью команды Load необходимо указать имя файла, в котором он хранится (предварительно его надо записать в текущий каталог). При удалении какого-либо спектра из списка с помощью команды Deleteсоответствующий ему файл не удаляется, поэтому его всегда можно будет включить обратно в список с помощью команды Load.
Создание и редактирование групп базисных спектров
. В программе STRUCTUREуже существует 6 предопределенных групп базисных спектров, соответствующих различным методам анализа спектров КД. Эти группы имеют следующие имена:
- PG_3_16.GRPи PG_4_16.GRP - базисные наборы, состоящие из 16 спектров, использованные для анализа авторами метода "регуляризации" [4] (Provencher& Glockner), предназначенные для определения вторичной структуры по 3 и 4 структурным классам соответственно (смотри ниже);
- PG_3_20.GRPи PG_4_20.GRP - базисные наборы, содержащие те же самые 16 спектров, что и в двух предыдущих наборах, плюс 4 спектра денатурированных белков;
- HJ_16.GRPи HJ_22.GRP - базисные наборы, состоящие из 16 и 22 спектров соответственно, использованные для анализа авторами метода "ортогональных спектров" [7] (Henessey& Johnson), предназначенные для определения вторичной структуры по 5 структурным классам (смотри ниже).
В программе предусмотрена возможность создания собственных групп базисных спектров. Для этого необходимо воспользоваться командой главного меню Group/Create, позволяющей выбрать из списка существующих спектров те, которые вы хотите включить в свой базисный набор. Аналогичным образом осуществляется редактирование групп базисных спектров (команда главного меню Group/Edit). Удаление группы базисных спектров осуществляется с помощью команды главного меню Group/Delete.
Выбор набора
структурных типов
. В программе STRUCTUREпредопределены следующие 3 набора типов вторичной структуры белка:
Provencher 3 (PG3.STR)
|
ALFA_hl (a-спираль)
BETA_sh (b-структура)
Remain (остальные типы)
|
| Provencher 4 (PG4.STR) |
ALFA_hl (a-спираль)
BETA_sh (b-структура)
BETA_tn (b-поворот)
Remain (остальные типы)
|
| Johnson 5 (HJ.STR) |
ALFA_hl (a-спираль)
BETA_Ash (антипараллельная b-структура)
BETA_Psh (параллельная b-структура)
BETA_tn (b-поворот)
Other (остальные типы)
|
Набор Allstructures (FULL.STR) содержит дополнительные типы вторичной структуры белка, однако он ни с одной из предопределенных групп базисных спектров не используется.
Каждая группа базисных спектров соответствует одному из выше перечисленных наборов структурных типов. Это соответствие выглядит следующим образом:
PG_3_16.GRPи PG_3_20.GRP - Provencher3;
PG_4_16.GRP и PG_4_20.GRP - Provencher 4;
HJ_16.GRP и HJ_22.GRP - Johnson 5.
При выборе одной из групп базисных спектров необходимо выбрать соответствующий набор типов вторичной структуры белка. Выбор нужного набора структурных типов осуществляется с помощью команды главного меню Options/Structuretypes.
Запуск вычислений
. Для начала вычислений необходимо воспользоваться командой главного меню Calculate. В появляющемся меню нужно выбрать один из предлагаемых методов вычислений. В появляющемся после этого списке имеющихся белковых спектров необходимо выбрать анализируемый спектр. Если для расчетов были выбраны программы CONTIN, VARSELEC, PROVCDили DEF_CLASS, то необходимо также выбрать группу базисных спектров, на которой будут основаны вычисления. После этого производится запуск вычислений.
Если для расчетов была выбрана программа VARSELEC, то необходимо также установить порядок исключения спектров из исходного базисного набора для процедуры "выбора переменных" с помощью команды главного меню Options/Var.select. Для этого необходимо указать число спектров, исключаемых на каждом шаге вычислений. После его задания автоматически вычисляется общее количество шагов, требуемых для перебора всех возможных комбинаций. Если перебор всех возможных комбинаций не требуется, необходимо указать номер начальной и конечной комбинации.
Время вычислений равняется в среднем 1-3 минутам, однако может составлять значительно больший интервал для программы VARSELECпри задании очень большого количества комбинаций базисных спектров.
Результаты вычислений можно просмотреть с помощью команды Calculate/Result.
Поглощение света в видимом и ультрафиолетовом диапазонах обусловлено электронными переходами в молекулах поглощающего вещества. Поглощение света в инфракрасном диапазоне имеет иную природу. Оно связано с переходами между колебательными уровнями основного состояния молекулы. Полосы поглощения, отвечающие колебательным переходам, обычно лежат в диапазоне длин волн от 2000 до 50000 нм или, как принято записывать для ИК-спектров, в диапазоне волновых чисел от 5000 до 200 см-1
.
Колебательные спектры подчиняются в сущности тем же закономерностям, что и электронные. Однако для колебательных переходов характерна значительно меньшая интенсивность, чем для электронных. Следовательно, при регистрации ИК-спектра образец должен быть гораздо более концентрированным. Кроме этого, многие полосы ИК-спектров белков, в том числе соответствующие пептидным хромафорам, расположены в той спектральной области, где наблюдается сильное поглощение воды. Использование D2
О вместо Н2
О иногда помогает обойти эту трудность, но не решает проблему полностью, поскольку полная замена лабильных протонов белка на дейтерий часто связана с потерей его нативной конформации.
Описываемый ниже метод определения вторичной структуры белка основан на использовании ИК-спектров поглощения белков в Н2
О [8-10]. Проблема, связанная с их измерением, была решена авторами метода с помощью довольно сложной процедуры компенсации поглощения воды и использования очень узких ячеек (с длиной оптического пути около 6-12 мкм). Поскольку все измерения проводились в Н2
О трудностей с поддержанием нативной конформации белков не возникало.
Колебательные полосы поглощения обычно порождаются переходами, которые можно довольно точно отнести к определенным химическим связям. В случае белков наиболее интересными являются три инфракрасные полосы, соответствующие колебательным переходам в пептидном остове. Это полосы, связанные с растяжением связи N-H (около 3300 см-1
), растяжением связи C=O (1640-1660 см-1
, полоса амид I) и деформацией связи N-H (1520-1550 см-1
, полоса амид II). Эти полосы довольно легко зарегистрировать, поскольку каждое пептидное звено дает вклад в их интенсивность.
Образование водородных связей при формировании вторичной структуры белка приводит к сдвигу энергии этих трех пептидных колебаний. Первые две полосы, отвечающие валентным колебаниям, смещаются в область более низких энергий, поскольку наличие водородной связи облегчает смещение атома азота амидной группы и атома кислорода карбонильной группы в направлении акцептора или донора протона соответственно. Полоса амид IIсмещается в сторону более высоких энергий, так как водородная связь препятствует изгибанию связи N-H.
Влияние водородных связей на полосы амид Iи амид IIв случае a-спирали и b-структуры оказывается различным, что дает возможность использовать ИК-спектры для определения вторичной структуры белков. Ниже представлена таблица, суммирующая данные о влиянии вторичной структуры на положение полос амид Iи амид II. В ней приведены положения максимумов (n0
) и значения интенсивности в максимумах (Е0
) для полос амид Iи амид II, усредненные по нескольким модельным полипептидам и фибриллярным белкам в Н2
О [9]:
| Тип вторичной |
Амид I |
Амид II |
| структуры |
n0
, см-1
|
Е0
, л·моль-1
·см-1
|
n0
, см-1
|
Е0
, л·моль-1
·см-1
|
a-спираль
|
1647 |
700 |
1551
1520
|
310
80
|
b-структура
|
1695
1619
|
180
980
|
1533
1563
|
340
100
|
неупорядочен-
ная форма
|
1651 |
320 |
1550 |
210 |
Следует отметить, что расщепление полос амид Iи амид IIпроисходит за счет взаимосвязанности колебаний в отдельных пептидных группах.
На рисунке 2.1.1 представлены ИК-спектры трех модельных полипептидов, находящихся в конформациях a-спирали, b-структуры и неупорядоченной формы.
В целом, проблемы, решаемые при анализе ИК-спектров белков с целью определения их вторичной структуры, очень схожы с проблемами, возникающими при анализе спектров кругового дихроизма белков. При этом также используется набор ИК-спектров белков с известной вторичной структурой, используемых в качестве базисных. Так, например, в методе, описанном в работах [8-10], анализ базисного набора, состоящего из 13 спектров глобулярных белков и 6 спектров фибриллярных белков и полипептидов в Н2
О в диапазоне 1800-1480 см-1
, осуществляется с помощью методов "регуляризации" [4] и "ортогональных спектров" [6], рассмотренных выше.
Авторы этого метода вводят дополнительную процедуру, позволяющую исключить вклад в ИК-спектр белка поглощения боковых групп аминокислотных остатков. Ими было показано, что этот вклад составляет около 20% от суммарной интенсивности полос амид Iи амид II. Возможность проведения такой процедуры определяется тем, что вклады в ИК-спектр поглощения белка от боковых групп аминокислотных остатков и полипептидного остова являются аддитивными. Для оценки поглощения боковых групп было проведено измерение ИК-спектров водных (Н2
О) растворов аминокислот. Оказалось, что наиболее сильно поглощают в исследуемой части ИК-диапазона боковые группы аминокислот аспарагина, глутамина, аспарагиновой кислоты, глутаминовой кислоты, аргинина, лизина, тирозина, фенилаланина и гистидина. Было обнаружено также сильное поглощение заряженных a-амино - и a-карбоксильной групп аминокислот. Поэтому их поглощение также необходимо учитывать при анализе белкового спектра. Суммарно, ИК-спектр белка может быть представлен в следующем виде:
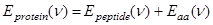 . (2.2.1) . (2.2.1)
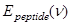 - спектр поглощения полипептидного остова белка, а - спектр поглощения полипептидного остова белка, а 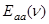 - спектр поглощения боковых групп аминокислотных остатков белка, вычисляемый по формуле - спектр поглощения боковых групп аминокислотных остатков белка, вычисляемый по формуле
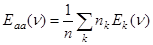 , (2.2.2) , (2.2.2)
где 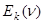 - спектр поглощения k
-ой аминокислоты, - спектр поглощения k
-ой аминокислоты, 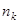 - число k
-ых аминокислот в белке, а - число k
-ых аминокислот в белке, а 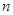 - общее число аминокислот в белке. N - и С-концевые - NH2
и - COOHгруппы белка также должны быть включены в эту формулу наравне с аминокислотами. Пример исключения из ИК-спектра рибонуклеазы А вклада от поглощения боковых групп аминокислотных остатков приведен на рисунке 2.2.1 Таким образом, данный метод анализа ИК-спектров полностью аналогичен методам анализа спектров КД белков, за исключением того, что в нем используются не реальные белковые спектры, а вычисленные с помощью формул (2.2.1) и (2.2.2) спектры поглощения пептидного остова белков. - общее число аминокислот в белке. N - и С-концевые - NH2
и - COOHгруппы белка также должны быть включены в эту формулу наравне с аминокислотами. Пример исключения из ИК-спектра рибонуклеазы А вклада от поглощения боковых групп аминокислотных остатков приведен на рисунке 2.2.1 Таким образом, данный метод анализа ИК-спектров полностью аналогичен методам анализа спектров КД белков, за исключением того, что в нем используются не реальные белковые спектры, а вычисленные с помощью формул (2.2.1) и (2.2.2) спектры поглощения пептидного остова белков.
Авторы метода использовали для анализа шесть типов вторичной структуры белка: упорядоченная (Ho
) и неупорядоченная (Hd
) формы a-спирали, упорядоченная (Вo
) и неупорядоченная (Вd
) формы b-структуры, b-изгиб (Т) и остальные формы (R). К неупорядоченной форме a-спирали были отнесены по два аминокислотных остатка с каждой стороны спирального сегмента, а к неупорядоченной форме b-структуры - аминокислотные остатки b-нитей, образующие "неклассические" водородные связи.
Применение к выбранному базисному набору метода "ортогональных спектров" [6] привело к получению 11 ортогональных спектров, амплитуда которых превышает экспериментальную ошибку, возникающую при регистрации ИК-спектров. Пять "наиболее значимых" ортогональных спектров показано на рисунке 2.2.2.
Экспериментальная проверка точности анализа ИК-спектров белков на белках из базисного набора дала следующие коэффициенты корреляции (смотри раздел 1.2):
| Метод |
Ho
|
Hd
|
Bo
|
Bd
|
T |
R |
| "регуляризации" |
0.97 |
0.77 |
0.94 |
0.91 |
0.80 |
0.48 |
| "ортогональных спектров" |
0.98 |
0.80 |
0.93 |
0.90 |
0.75 |
0.48 |
| "регуляризации" (без исключения поглощения боковых групп аминокислот) |
0.92 |
0.68 |
0.85 |
0.90 |
0.53 |
0.32 |
Пакет программ STRUCразработан в институте белка РАН [10]. Он предназначен для анализа инфракрасных спектров поглощения белков и определения их вторичной структуры. Алгоритм анализа спектров основан на оригинальном методе авторов программы [8-10] (смотри выше). Пакет STRUCсостоит из следующих программ и вспомогательных файлов:
- STRUC.BAT - командный файл, используемый для определения вторичной структуры белка. Он организует работу следующих трех программ:
- AMACIR (файл amacir.exe) - программа, исключающая из ИК-спектра поглощения белка вклад от поглощения боковых групп аминокислотных остатков по методу [8];
- SVDIR (файл svdir.exe) - программа, определяющая вторичную структуру белка методом "ортогональных спектров" [6];
- CONTIR (файл contir.exe) - программа, определяющая вторичную структуру белка методом "регуляризации" [4].
- VISUAL.BAT - командный файл, предназначенный для графического воспроизведения ИК-спектра поглощения белка. Он организует работу следующих двух программ:
- PLOTFL (файл plotfl.exe) - программа, осуществляющая преобразование спектров поглощения к формату, используемому программой PLOTNIK;
- PLOTNIK (файл plotnik.exe) - программа, осуществляющая графическое построение ИК-спектра поглощения белка.
- SOURCE - файл, содержащий входные данные для программы AMACIR (в том числе сам ИК-спектр поглощения белка).
- OUT.SVDи OUT.CON - файлы, являющиеся результатом работы программ SVDIRи CONTIR, содержащие данные по оценке вторичной структуры белка соответствующими методами.
Программы, входящие в пакет STRUC, не имеют системы экранного интерфейса, и работа с ними осуществляется из командной строки. Перед началом работы с пакетом необходимо создать текстовый файл SOURCE (рекомендуется скопировать уже имеющийся) и записать в него исходные данные по исследуемому белку и его ИК-спектру поглощения, необходимые для работы программ. Формат этого файла следующий:
| Номер строки |
Содержание |
| 1 |
Идентификатор файла (длиной не более 70 символов) |
| 2, 3 |
Формат записи числовых значений спектра в строках 6 - 14. По умолчанию устанавливается формат (10F6.0) - по 10 значений в каждой строке в форме действительного числа с фиксированной десятичной точкой, причем на запись всего числа отводится 6 позиций, из которых 0 позиций - на десятичную часть. (Для действительных чисел с плавающей десятичной точкой вместо символа 'F' следует записать символ 'E'.) |
| 5 |
Спектральный диапазон (по волновым числам), используемый для вычислений, и число точек в спектре. Максимально допустимый спектральный диапазон составляет 1800 - 1480 см-1
при числе точек в спектре, равном 81. При задании диапазона и числа точек необходимо сохранять интервал между точками равным 4 см-1
. |
| 6 - 14 |
ИК-спектр поглощения белка, выраженный в единицах л моль-1
·см-1
. |
| 17 |
Содержит единственный символ '*', положение которого определяет тип записи данных по аминокислотному составу белка: (а) NAmAc - указывается абсолютное количество остатков каждой аминокислоты в белке; (б) NAmAc/N - указывается абсолютное количество остатков каждой аминокислоты в белке, деленное на общее количество аминокислотных остатков. |
| 19 - 40 |
Аминокислотный состав белка. Здесь же отдельно указываются N - и C-концевые группы белка. |
| 42 |
Общее количество аминокислотных остатков в белке и значение pH, при котором снимался спектр. Указывать значение pHнужно обязательно, а общее количество аминокислотных остатков - только в том случае, если при записи аминокислотного состава белка записывались относительные доли аминокислотных остатков (вариант (б)). |
| 43 |
Комментарий к файлу |
Пример заполнения файла можно посмотреть в уже имеющемся файле SOURCE.
После подготовки файла SOURCEнеобходимо запустить вычисления. Для этого в командной строке следует ввести
STRUC SOURCE OUT.SVD OUT.CON и нажать кнопку [Enter]. (Названия файлов SOURCE, OUT.SVDи OUT.CONмогут быть любыми, следует лишь соблюдать указанный порядок пр их записи.)
После окончания работы программ результаты вычислений моут быть просмотрены в файлах OUT.SVDи OUT.CONс помощью любого текстового редактора. Первая часть этих файлов содержит информацию о входных данных (взятую из файла SOURCE). Далее следует набор решений, соответствующий различным комбинациям ортогональных спектров (в файле OUT.SVD) или различным значениям параметра регуляризации (в файле OUT.CON). В конце файлов приводится аппроксимированный спектр и окончательно выбранное решение.
Можно просмотреть также промежуточные результаты вычислений программ. Файл PEPT, создаваемый программой AMACIR, содержит ИК-спектр поглощения пептидной цепи. Этот файл является входным для программ SVDIRи CONTIR. Файл AMACIR.RES, также создаваемый программой AMACIR, содержит данные об ионизации отдельных аминокислотных остатков при заданном значении pH, среднем значении массы одного аминокислотного звена данного белка, содержании в белке азота, а также три ИК-спектра поглощения: спектр белка (определенный экспериментально) и спектры боковых групп аминокислотных остатков и пептидной цепи (вычисленные программой AMACIR).
Для того, чтобы построить все три спектра на экране, необходимо запустить файл VISUAL.BAT.
В появляющемся после этого на экране главном меню программы PLOTNIKследует выбрать пункт Suit, позволяющий перейти в раздел задания параметров, необходимых для построения графиков. Ниже перечислены основные из них:
Titles/X
Titles/Y
Scale/X/Upper
Scale/X/Lower
Scale/Y/Upper
Scale/Y/Lower
Misc/Npts
Misc/Data
|
Волновое число, 1/см
Коэффициент молярной экстинции, 1/ (моль см)
1480
1800
Auto
Auto
Auto
Formatted
|
Для одновременного построения трех графиков необходимо задать три набора данных (A, Bи C) и указать имена файлов, содержащих эти данные:
| Set |
Legend |
Filename |
A
B
C
|
экспериментальный спектр
спектр поглощения пептидной цепи
спектр поглощения а/к остатков
|
buf1.dat
buf2.dat
buf3.dat
|
Файлы buf1.dat, buf2.dat и buf3.dat создаются программой PLOTFL на основе данных файла AMACIR.RES (эти файлы уничтожаются после завершения работы программы PLOTNIK). После задания параметров следует вернуться в главное меню программы PLOTNIKи выбрать в нем пункт View. При этом на экране будут построены требуемые графики.
1. Кантор Ч., Шиммел П. Биофизическая химия. Том 2: Методы исследования структуры и функции биополимеров. М: Мир, 1984
2. Saxena V.P., Wetlaufer D.B. (1971) A new basis for interpreting the circular dichroic spectra of proteins. Proc. Natl. Acad. Sci. U.S.A.6
8, 969-972
3. Сhang C.T., Wu C. -S.C., Yang J.T. (1978) Circular dichroic analysis of protein conformation: inclusion of the b-turns. Anal. Biochem.9
1, 13-31
4. Provencher S.W., Glockner J. Estimation of globular protein secondary structure from circular dichroism. (1981) Biochemistry
20, 33-37
5. Hennessey J.P., Jr., Johnson W.C., Jr. (1981) Information content in the circular dichroism of proteins. Biochemistry
20, 1085-1094
6. Compton L.A., Johnson W.C., Jr. (1986) Analysis of protein circular dichroism spectra for secondary structure using a simple matrix multiplication. Anal. Biochem.1
55, 155-167
7. Manavalan P., Johnson W.C., Jr. (1987) Variable selection method improves the prediction of protein secondary structure from circular dichroism spectra. Anal. Biochem.1
67, 76-85
8. Venyaminov S.Yu., Kalnin N.N. (1990) Quantitative IR specrtophotometry of peptide compounds in water (H2
O) solutions.I. Spectral parameters of amino acid residue absorption bands. Biopolymers
30, 1243-1257
9. Venyaminov S.Yu., Kalnin N.N. (1990) Quantitative IR specrtophotometry of peptide compounds in water (H2
O) solutions. II. Amide absorption bands of polypeptides and fibrous proteins in a-, b-, and random coil conformations. Biopolymers
30, 1259-1271
10. Kalnin N.N., Baikalov I.A., Venyaminov S.Yu. (1990) Quantitative IR specrtophotometry of peptide compounds in water (H2
O) solutions. III. Estimation of the protein secondary structure. Biopolymers
30, 1273-1280
|
