| Федеральное агентство по образованию
Государственное образовательное учреждение высшего профессионального образования
Самарский государственный технический университет
Кафедра: «Органическая химия»
ПРОИЗВОДНЫЕ ИЗОКСАЗОЛЫ:
ПОЛУЧЕНИЕ, СВОЙСТВА И ПРИМЕНЕНИЕ.
СИНТЕЗ 5-НИТРОБЕНЗИМИДАЗОЛА
Курсовая работа
Выполнил: студент
_________________
(подпись)
Руководитель:
_________________
(подпись)
Работа защищена
“___“ __________ 2007г.
Оценка _______
Зав. кафедрой: доцент, д. х. н.
_________________
(подпись)
Самара, 2007 г.
Содержание
Содержание..................................................................................................... 2
1. Введение..................................................................................................... 3
1.1. Общие сведения........................................................................................ 3
1.2. Применение.............................................................................................. 4
2. Обзор литературы. Производные изоксазола......................................... 5
2.1. Общие сведения.................................................................................. 5
2.2. Регио- и стереоконтроль в нитрилоксидном синтезе изоксазолов и 2-изоксазолинов................................................................................................. 5
2.2. Реакции модификации производных изоксазола................................. 11
2.3. Реакции модификации производных изоксазола................................. 14
2.3.1. Расщепление основаниями.................................................................. 14
2.3.2. Восстановительное расщепление изоксазолов и 2-изоксазолинов... 16
2.3.3.Восстановительное расщепление 2-изоксазолинов в β-оксикетоны 17
2.3.4. Восстановительное расщепление 2-изоксазолинов в γ-аминоспирты. 22
2.3.5. Восстановительное расщепление изоксазолов............................ 25
3. Методы синтеза........................................................................................ 28
4. Выводы..................................................................................................... 29
Список литературы....................................................................................... 30
Реклама

1.1. Общие сведения
3,5-Диметилизоксазол – прозрачная от бесцветного до слегка желтоватого цвета жидкость. Содержание более 98,0% вода менее 0,5 %. Легко воспламеняемое вещество.
3,5-Диметилизоксазол – производное изоксазола. 1,2-Оксазол – бесцветная жидкость с запахом пиридина, темп. кипения 95,5оС, ограниченно растворимая в воде (1 масс. ч. в 6 масс. ч. воды), хорошо растворима в органических растворителях. Протонируется сильными кислотами по атому азота.
Под действием оснований депротонируется по атомам С-3 и С-5 с разрывом связи N—О и образованием α-кетонитрила. Обладает ароматическими свойствами. Для изоксазола характерно электрофильное замещение (нитрование, сульфирование, галогенирование) по атому С-4, для производных изоксазола – нуклеофильное замещение по атомам С-3 и С-5. Алкилирование изоксазола протекает по атому N. Изоксазол устойчив к действию окислителей, кроме КМnО4; восстановление приводит к разрыву цикла по связи N—О. Под действием света изоксазол и его гомологи изомеризуются сначала в ацилазирины, затем в оксазолы, например:
Изоксазол получают взаимодействием гидроксил амина с пропаргиловым альдегидом 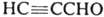 или его ацеталем. Производные изоксазола синтезируют взаимодействием гидроксиламина с β-дикарбонильными соединениями, а также циклоприсоединением N-оксидов нитрилов к алкинам, напр.: или его ацеталем. Производные изоксазола синтезируют взаимодействием гидроксиламина с β-дикарбонильными соединениями, а также циклоприсоединением N-оксидов нитрилов к алкинам, напр.:
Производные изоксазола широко используют в органической химии для получения α,β-ненасыщенных кетонов, α-кетонитрилов, гетероциклических соединений, а также в синтезе лекарственных средств (например, циклосерина, оксациллина, клоксациллина). Так, производные изоксазолы применяются при лечении и профилактике ревматоидного артрита.
Растущий интерес к химии производных изоксазола и к использованию их в синтезе природных соединений и их аналогов, включая простаноиды, антибиотики, противоопухолевые вещества, витамины, нуклеозиды и алкалоиды. Тем не менее прогресс, достигнутый в этой области в последние годы, делает необходимым дальнейшую систематизацию информации о получении и химических превращениях изоксазолов и 2-изоксазолинов. Образование изоксазольного цикла и его расщепление происходят с определенной регио- и стереоселективностью; синтезы через производные изоксазола являются стереоконтролируемыми. Это очень важно в полном синтезе соединений, имеющих несколько хиральных центров.
Реклама

Гетероциклические соединения, обладающие латентной функциональностью, широко используются в органическом синтезе. Весьма показательны в этом плане изоксазолы и их 1,2-дигидропроизводные — 2-изоксазолины. Благодаря интенсивной разработке «нитрилоксидной» технологии они являются доступным и эффективным средством построения углеродного скелета органических соединений различных классов. При этом реализация латентной функциональности изоксазольного ядра путем расщепления гетероцикла дает выход к таким важным соединениям, как р-дикетоны, енаминокетоны, еноны, р-оксикетоны, еноксимы, Y-аминоспирты и др.
В синтезе природных соединений и их аналогов производные изоксазола используют для построения и/или удлинения углеродной цепи конструирования полициклической молекулы, а также для функционализации олефиновых фрагментов молекулы. Стратегия изоксазольного (нитрилоксидного) метода синтеза органического соединения (или его фрагмента) состоит из трех этапов: 1) синтез гетероцикла I реакцией 1,3-диполярного циклоприсоединения нитрилоксида II in situ (при дегидратации нитроалкана или дегидрохло-рировании хлорида оксима) к непредельному соединению; 2) модификация молекулы I введением алкильных заместителей или функциональных групп либо в цикл, либо в экзоциклическое положение; 3) раскрытие цикла, приводящее к бифункциональному производному III.
Первый этап – 1,3-диполярное циклоприсоединение – может быть осуществлен внутримолеклярно (ненасыщенная связь С-С – диполярофил и нитрилоксид — диполь являются частями одной молекулы) и межмолекулярно (гетероцикл формируется из двух разных молекул — молекулы-диполя и молекулы-диполярофила). Межмолекулярное циклоприсоединение применяют для конвергентного синтеза природных соединений из готовых блоков, содержащих необходимые функции (или их эквиваленты) в заместителях R1 и R2.
Доступность производных изоксазола различного строения обеспечивается практически неограниченным диапазоном реакции 1,3-диполярного циклоприсоединения, протекающей в мягких условиях с высоким выходом циклоаддуктов из разнообразных непредельных соединений и предшественников нитрилоксидов. Важным преимуществом циклоприсоединения является его цис-стереоспецифичность. Проблемы селективности нитрилоксидного синтеза возникают из-за возможности образования в реакции двух региоизомерных изоксазолов или 2-изокса-золинов. Кроме того, в реакции с алкенами подход диполя (нитрилоксида) к диполярофилу может происходить с обеих сторон от плоскости двойной связи, поэтому можно ожидать образование диастереомерной пары а, б изоксазолинов, а предпочтение какой-либо из сторон для атаки приводит к более или менее заметной диастереоселективности.
Проведение синтеза в стереоконтролируемых условиях уже на первом этапе изоксазольного метода является предпосылкой высокого выхода стереохимически однородных интермедиатов и конечных продуктов синтеза.
Основными факторами, определяющими региоселективность нитрилоксидного синтеза, являются степень поляризации ненасыщенной системы и объем заместителей R2 и R3. При этом кислород нитрилоксида связывается с более положительно заряженным и стерически затрудненным концом двойной или тройной связи С—С. Это правило хорошо выполняется в частном случае монозамещенных, или «терминальных», непредельных соединений; при переходе к неактивированным олефинам и ацетиленам стерические факторы играют решающую роль. Факторы, контролирующие диастереоселективность реакции, установлены при исследовании присоединения различных нитрилоксидов к производным З-бутен-2-ола и 3-бутен-1,2-диола (VI—XVI). В этом случае теоретической предпосылкой возможности контроля диастереоселективности была концепция антиперипланарного присоединения, в соответствии с которой нитрилоксид подходит к связи С—С со стороны, противоположной аллильному заместителю OR; при этом сводятся к минимуму несвязывающие взаимодействия кислорода с R1 нитрилоксида, а атом кислорода в аллильном положении занимает наиболее выгодную ортогональную ориентацию к плоскости связи С—С.
Однако из экспериментальных данных следует, что это предположение выполняется только для цис-замещенных олефинов, например соединения XVI. Для терминальных и гранс-дизамещенных олефинов нужно рассматривать не только переходное состояние (А) с ортогональным атомом кислорода в аллильном положении, но и кон-формацию (В) с наклоном заместителя к плоскости связи С=С, с помощью которой можно объяснить заметную селективность присоединения в этих случаях (XIV, XV). Ориентирующее влияние «аллильного» кислорода проявляется в совокупности с другими факторами строения аллильного заместителя. Так, необходимо учитывать объем заместителя при связи С==С (ср. VI, IX, XI, XIV) и наличие аллильного хирального центра, вызывающего асимметрическую индукцию. При присоединении бензонитрилоксида к хиральным олефинам соотношение диастерео-изомерных изоксазолинов меняется в зависимости от строения олефина, достигая значительной величины при наличии пятичленного цикла в аллильном положении (ср. VI, IX, XIV). По данным расчетов моделей переходного состояния конформация с антиперипланарным расположением группы СН2Х предпочтительна для любого — эритро- или трео-диастереомера, поскольку в этом случае не сказывается стерический эффект заместителя X. Вопрос, однако, состоит в том, как расположен заместитель Y — «внутри», как в эритроизомере, или «снаружи», как в тpeoизомере. При увеличении объема заместителя Y резко меняется стереохимический результат (ср. XII и XIII, табл. 1): возрастает количество более выгодного эритроизомера, поскольку стерический фактор Y в тpeoконформации должен сказываться больше.
Стереохимический результат внутримолекулярного циклоприсоединения определяется совокупностью многих факторов строения субстрата. Для монозамещенных терминальных алкенов стереоселективность контролируется напряжением формирующейся бициклической системы..
Отмеченный для межмолекулярного циклоприсоединения амты-ориен-тирующий эффект аллильного асимметрического центра с объемнымзаместителем вблизи него при внутримолекулярном циклоприсоединении не проявляется в заметной степени. Согласно данным расчетов моделей переходного состояния реакции для Z-алкенов предпочтительна конформация Х1Ха с расположенным «внутри» по отношению к образующейся связи С—О наименьшим по объему заместителемаллильного хирального центра; диастереоселективностью управляют главным образом стерические факторы. Для имеющей меньше стерических ограничений двойной связи Е-алкенов предполагают, что «внутри» находится средняя по объему группа Y, поскольку наблюдается зависимость стереоселективности от электронных факторов заместителей Y и X.
При этом, если одним из заместителей аллильного стереоцентра является гетероатом, стереоселективность внутримолекулярного циклоприсоединения резко возрастает. Так, высокую стереоселективность, показанную аллиловыми эфирами на основе глицеринового альдегида, связывают именно с электронными факторами обоих — аллильного и гомоаллильного атома кислорода. Таким образом, влияние алкоксильной группировки Y у аллильного стереоцентра («inside alkoxy effect») на стереоселективность как меж-, так и внутримолекулярного циклоприсоединения, очевидно, обусловлено как электронным фактором гетероатома, так и объемом всей группы RO.
В заключение следует отметить, что в стерерконтроле нитрилоксидного синтеза решающую роль играет строение олефина. Описан ряд случаев; когда присоединение нитрилоксидов весьма сложного строения к простым олефинам протекает без заметной селективности и лишь при использовании оптически активных нитрилоксидов наблюдается некоторый перенос хиральности. Поэтому нитрилоксиды рассматривают как относительно малые циклоадденды, и только для объемного и. оптически активного нитрилоксида можно предположить, что циклоприсоединение будет происходить с тем большей стереоизбирательностью, чем больше будет условий для осуществления стереоизбирательности в конкретном олефине.
Использование информации о факторах стереоконтроля нитрилоксидного синтеза дало возможность; успешно осуществить стереоселективные синтезы изоксазолиновых предшественников 2-дезокси-Б-рибозы, ключевого интермедиата в синтезе углеводов — «компактинлактона», метаболита антибиотика антимицина — бластмицинона и других природных соединений.
Изоксазольный цикл устойчив к действию многих обычно используемых в синтезе реагентов — сильных кислот, мягких восстановителей, сильных окислителей. Положительный аспект латентной функциональности изоксазольного ядра состоит в том, что в различные положения молекулы можно ввести функциональные группировки или модифицировать уже имеющиеся, не затрагивая сам гетероцикл. При этом малый геометрический размер и компактность гетероцикла не создают препятствий для проведения реакций.
Основной путь модификаций 2-изоксазолинов базируется на их способности вступать в реакции замещения. При действии сильных оснований происходит отрыв либо одного из аллильных протонов при атоме С(4) цикла (4-эндо-депротонирование), либо в заместителе при С(3) цикла (3-экзодепротонирование) с образованием стабильного при -60—80°С аниона, который может взаимодействовать с различными электрофилами. Так, 3,5-дифенилизоксазолин XX при действии диизопропиламида лития (LDA) в ТГФ при - 78 °С образует 4-экзо-анион (С), алкилирование которого происходит транс-стереоселективно по отношению к заместителю при С(5). Этот метод позволяет получать 4-транс-R-изоксазолины XXI, которые не всегда доступны реакцией нитрилоксидного присоединения к транс-алкенам из-за ее низкой селективности. Потенциальные предшественники аминосахаров — 4-гидроксиизоксазолины XXII — недоступны нитрилоксидным синтезом, поскольку в циклоприсоединении заместитель OR алкена занимает положение 5 гетероцикла, но их также можно получить методом транс-селективного 4-эндо-гидроксилирования.
Атом водорода при третичном атоме С(4) в 4-метилизоксазолине XXI (Е=Ме) может снова отщепляться, благодаря чему возможно получение 4-гем-диметилизоксазолина. Для 3-алкилзамещенных изоксазолинов было установлено, что алкилирование заместителя при С(3) идет после алкилирования цикла, т. е. 4-эндопротон имеет более высокую кинетическую кислотность и депротонируется первым. Для 3,4,5-тризамещенных изоксазолинов, в частности для 3-алкил-4,5-цикло-пентаноизоксазолинов, предпочтительное 3-экзо-алкилирование объясняется меньшей кинетической кислотностью эндометинового водорода по сравнению с экзометильным водородом. Региоселективность депротонирования зависит, однако, от используемого растворителя: в неполярных растворителях наблюдается региоспецифическое 3-экзо-депротонирование. Значительное увеличение региоселективности достигается при использовании более объемного литийамидного основания.
Факторы стереоселективности эндоалкилирования гетероцикла были изучены на примере изоксазолинов XXIII и установлено, что кислородсодержащий заместитель при атоме С(5) направляет алкильный заместитель преимущественно в транс-положение. Предполагается, что в реакции образуется переходный комплекс (D), в котором кислород заместителя OR при С(5) хелатируется с катионом лития, координированным с 4-эндоанионом, тем самым син-сторона этого комплекса закрывается для атаки электрофильной частицей. Таким образом обеспечивается предпочтительность введения новой алкильной группы напротив OR, даже в случае 4-метил-5-алкоксиизоксазолина.
Основным фактором стереоконтроля 3-экзо-алкилирования являете; заместитель при атоме С(4) изоксазолина, по отношению к которому замещение идет преимущественно транс-стереоселективно.
Депротонирование 3,5-диметилизоксазолов происходит региоизбирательно сначала по метальной группе при атоме С(5), а затем по метилу при С(3), так что при последовательном замещении можно получит! различные 3,5-дизамещенные изоксазолы.
Подвижность аллильных протонов в положениях 3 и 5 изоксазол; и положениях 4 и 5 изоксазолина может быть использована для введения различных функциональных групп. Например, 3,5-диметил-4-нитроизоксазол использован в синтезе кумариновой кислоты i качестве СН-кислотного компонента реакции Перкина. При синтезе ланкацидйна разработан метод одностадийного последовательной ацилирования и алкилирования изоксазолинового цикла по атому С(4).
Синтетически полезные модификации можно проводить на основе галогензамещенных изоксазолинов и изоксазолов, которые получают нитрилоксидным синтезом с использованием α-галогензамещенных олефинов или нитрилоксидов. Такие производные изоксазола (XXIV, XXV) легко вступают в реакцию нуклеофильного замещения, обеспечивая выход к широкому кругу производных XXVI, имеющих различные функции в заместителях гетероциклического ядра.
Таким образом, возможность структурной модификации изоксазолов и 2-изоксазолинов расширяет применимость этих универсальных гетероциклов для синтеза большого числа полифункциональных молекул.
Реализация синтетического потенциала изоксазолов и их производных достигается раскрытием цикла под действием в основном двух типов реагентов — восстановителей и оснований.
Обобщая информацию большого числа исследований по расщеплению изоксазолов и 2-изоксазолинов основаниями, подробно изложенную в обзоре, можно утверждать, что получение однозначного результата проблематично из-за сильной зависимости направления раскрытия цикла от строения субстрата, основания и условий реакции. Многие реакции идут под действием одних оснований и не идут под действием других. Образующийся под действием оснований анион типа (С) (при комнатной температуре расщепляется, причем нравление и легкость раскрытия цикла зависят от строения производного изоксазола, поскольку именно строением определяется место депротонирования и его доступность для основания. У незамещенных по С(3) изоксазолов и изоксазолинов происходит раскрытие цикла по связи N—О с превращением в нитрилы и их производные XXVII, XXVIII. 3-Замещенные изоксазолины расщепляются по связи С—О с образованием еноксимов XXIX. Еноксимы далее могут быть превращены в α,β-еноны, восстановлены в амины или рециклизованы. 3-Замещенные изоксазолы расщепляются основаниями с образованием енаминокетонов XXX. В мягких условиях (0°С) происходит расщепление изоксазолиевых солей XXXI, поэтому такой вариант раскрытия цикла наиболее приемлем для лабильных производных изоксазола. Недавно предложен интересный препаративный метод расщепления изоксазолиниевых солей основаниями.
Как видно из схемы, ненасыщенность гетероцикла определяет как региохимию расщепления, так и степень окисленности продуктов раскрытия: 2-изоксазолины дают в качестве продуктов в основном еноксимы XXIX, а более прочный гетероаромэтический цикл раскрывается по связи N—О.
Общая схема гидрогенолиза 2-изоксазолинов и изоксазолов по связи N—О предполагает промежуточное образование либо оксиимина ХХХШ, либо кетоимина XXIV соответственно, а конечные продукты образуются в результате дальнейшего восстановления (путь а) или гидролиза (путь б) этих промежуточных. Оксиимины, долгое время считавшиеся гипотетическими интермедиатами, недавно были выделены и их строение доказано. Кетоимины вполне устойчивы.
Направление дальнейшего превращения оксиимина в аминоспирт XXXV или оксикетон XXXVI либо кетоимина в енаминокетон XXX или дикетон XXXII определяется природой восстанавливающего агента и условиями реакции. Образование аминоспирта при восстановлении 2-изоксазолинов. в некоторых случаях происходит через изоксазолидин. Образование других продуктов восстановления производных изоксазола в каждом частном случае обусловлено спецификой строения конкретного исходного соединения, которая проявляется либо на стадии раскрытия цикла, либо в дальнейших превращениях первичных продуктов гидрогенолиза.
Оксикетоны (альдоли) XXXVI являются первичными и основными продуктами гидрогенолиза изоксазолина и последующего гидролиза промежуточного оксиимина XXXIII. Для получения оксикетонов предложено довольно много методов, которые можно разделить, на две основные группы.
1. Методы каталитического гидрирования 2-изоксазолинов с использованием палладиевых и никелевых катализаторов. Среди них важное место принадлежит восстановительному расщеплению 2-изоксазолиновпри действии никеля Ренея в кислой среде. Впервые описанная в 1979 г. общая реакция расщепления изоксазолинов в β-оксикетоны или продукты их дегидратации — α,β-ненасыщенные кетоны впоследствии получила широкое распространение в различных методических модификациях. Считается, что в присутствии сильной кислоты обеспечивается стереоспецифичность раскрытия цикла. Наблюдаемое в случае 3,4,5-замещенных изоксазолинов нестереоспецифическое расщепление связано с тем, что при наличии объемного заместителя при атоме С(3) уменьшается скорость гидролиза оксиимина XXXIII и через таутомерное превращение последнего в енамин возможна эпимеризация при С(4).
При восстановлении на никеле Ренея изоксазолинов сложного строения оказалось, что в случае соединений, имеющих чувствительные к восстановлению заместители, большие преимущества имеет проведение реакции при значениях рН 5... 7; при этом расщепление не осложняется побочными процессами. Заслуживают внимания методические разработки с использованием борной кислоты и других соединений бора в качестве кислотных агентов, что гарантирует сохранность чувствительных к кислотам защитных групп — ацетильной, тетрагидропиранильной, силильной. Для сохранения чувствительных к восстановлению и кислотам групп предложена методика озонолитического расщепления изоксазолинов.
2. Применение экзотических восстанавливающих систем оправдано при наличии в изоксазолине непредельных заместителей, поскольку все методики гидрогенолиза с использованием никеля Ренея не обеспечивают сохранность непредельных группировок. В последнее время предложены новые восстановители — Мо(СО)6, Fe(CO)5,Н2/Rh-Al, имеющие существенные преимущества и отличающиеся большей селективностью действия.
Многочисленными исследованиями установлено, что расщепление изоксазолинов в оксикетоны происходит без обращения конфигурации и формально стереоспецифично от исходного олефина. Таким образом, геометрия олефина непосредственно транслируется в геометрию конечного алициклического соединения.
Такой стереоконтролируемый двустадийный способ получения альдольного фрагмента привлекателен для использования в органическом синтезе. Удобный путь к различным типам полифункциональных молекул открывает расщепление 3,4,5-функционально-замещенных изоксазолинов. Для их синтеза используют как нитрилоксиды, имеющие гидрокси-, алкокси-, циано-, алкоксикарбонильные заместители, так и α-функцйонализированные олефины. Так, разработаны синтезы β-оксикислот XXXVII из защищенных нитроспиртов через 3-алкоксиметилизоксазо-лины XXXVIII. Для этой цели также применяли бензолсульфонилнитрилоксид, в дальнейших превращениях бензолсульфогруппа легко заменяется на метоксигруппу. Недавно предложен более простой вариант синтеза — через 3-галогенизоксазолины XXV.
При использовании цианонитрилоксида и карбэтоксинитрилоксида разработаны методы получения оксинитрилов: 3-алкоксикарбонилизоксазолины XXXIX легко омыляются в 3-карбоксиизоксазолины, последние могут пиролизоваться с одновременным расщеплением в оксинитрилы XL.
Метод цис-циангидроксилирования предложен недавно и на основе 3-бензолсульфонилизоксазолинов.
3-Алкоксикарбонилизоксазолин XLI расщепляется диазометаном до Y-оксикислоты XLII, которая под действием трифторуксусной кислоты рециклизуется в лактон XLIII.
3-Изоксазолиновые кислоты XLIV под действием цинка в уксусной кислоте расщепляются по обычной схеме, однако присутствующая карбоксигруппа обусловливает циклизацию промежуточного оксиимина XLV в лактон XLVI, который затем восстанавливается в XLVII и ацилируется с образованием N-ацетиламинолактона XLVIII.
Разработан метод, с помощью которого из 3-гидроксиметилизокса-золинов XLIX через оксикетоны получают α-метиленлактоны.
Препаративные методы синтеза циклических кетодиолов LII и енкетолов LIII, являющихся ключевыми соединениями в полном синтезе стероидов, простаноидов и других биологически активных молекул, представляют собой пример изоксазольного метода функционализации циклоалканов.
В изоксазольных схемах синтеза простаноидных предшественников — метиленциклопентанонов ключевой стадией является расщепление из-оксазолинов LIV и LV гидрированием над Ni-Ra в мягких условиях с образованием оксикетонов и последующей дегидратацией их в α,β-не-предельные кетоны LVIa, LVI6.
Разработана схема синтеза функционализированных предшественников простаноидов LIX, LXII, в которой ключевыми реакциями являются образование и расщепление изоксазолинов LVII, LX и конденсация γ-кетоальдегидов LVIII или дикетонов LXI.
Аналогичный подход был применен для синтеза простаноидных синтонов исходя из диэтилацеталя акролеина. Изоксазольный метод генерирования оксикетонного фрагмента широко используется в синтезе других природных соединений и их аналогов. Образцовым примером использования всех этапов изоксазольной стратегии в. синтезе природных соединений является полный синтез бластмицинона, когда подбор субстратов и реагентов обеспечил проведение реакций в условиях стереоконтроля. При использовании нитрилоксида с α-асимметрическим центром и алкена с алкоксизаместителем в аллильном положении осуществлен стереоселективный синтез изо-сазолина, который после алкилирования был гидрогенолизом превращен в оксикетон с заданной стереохимией в α'-, α-, β- и -γ-центрах.
Гидрогенолиз изоксазолинов в аминоспирты — ключевая стадия синтеза многих природных соединений, таких, как оксиаминокислоты, моносахара и другие. Предполагают, что в зависимости от природы Восстанавливающего агента расщепление изоксазолина в аминоспирт может идти через оксиимин или изоксазолидин LXIII. Первый путь представляет собой гидрогенолиз цикла в оксиимин, дальнейшее восстановление которого приводит к аминоспирту XXXV. Второй путь предусматривает предварительное полное насыщение цикла, которое практически может быть осуществлено через его метилирование с образованием изоксазолиниевой соли и восстановлением в изоксазолидин LXIII гидридом металла. Для расщепления изоксазолидина используют амальгаму алюминия в водных растворах.
Подробно исследована стереохимия превращений замещенных изоксазолинов в аминоспирты, стереоселективность различных восстанавливающих агентов и другие факторы стереоконтроля реакции. Наилучшим восстановителем, как с точки зрения выхода аминоспирта, так и селективности, оказался алюмогидрид лития, способствующий «эритро»-селективности реакции.
Стереоселективность литийалюмогидридного восстановления заметно снижается при взаимном 4,5-грсшс-р'асположении заместителей в гетероцикле и увеличивается при наличии алкоксизаместителей при атоме С(5). При переходе от гидрокси- к алкоксизаместителю стереоселективность заметно возрастает (ср. LXVIII и LXXII; LXX и LXXI). Для алкильных и арильных заместителей при С(5) обнаружен анти-ориентирующий эффект, заместители с гидроксигруппой являются син-ориентантами. Маленький по объему реакционноспособный алюмогидрид лития чувствителен к строению и объему заместителей гетероцикла потому, что координируется за счет хелатирования лития и кислорода цикла, при этом алюминий и водород размещаются над связью С—N с наиболее доступной стороны и увеличивается общая анти-стереоселективность процесса восстановления. В субстратах, имеющих алкильные и фенильные заместители, анти-направление контролируется размером заместителя и можно предположить переходное -остояние (Е) для переноса водорода из алюминат-аниона. В случае субстратов с гидроксифункциями возникает дополнительный координационный комплекс, и гидридный перенос предпочтительнее из алкокси-алюминатного комплекса (F), поэтому заместители такого рода являются син-ориентантами.
Высокая диастереоселективность наблюдается при восстановлении изоксазолинов, имеющих при атоме С(5) диоксолановую группировку, а также фуро- и дигидрофуроизоксазолинов, в которых кислородсодержащий заместитель жестко закреплен в [3.3.0] бициклической системе. Исследована также степень асимметрической индукции заместителей цикла в условиях литийалюмогидридного восстановления и установлено, что гидроксиметильные заместители при атомах С(3) и С(5) уменьшают степень асимметрической индукции, при этом наибольшее влияние оказывает заместитель при С(5) [86, 90]. Сравнением эффектов заместителей при С(4) и С(5) также выявлено преобладающее влияние заместителя при С(5): 1,3-индукция преобладает над 1,2-индукцией. Диоксолановый заместитель, способствуя стереоселективному раскрытию цикла, вызывает уменьшение степени асимметрической индукции.
Исследование процесса последовательного восстановления через изоксазолидин показало, что восстанавливающие агенты средней силы (NaBH4) или очень реакционноспособные объемные агенты (L-селектрид) нечувствительны к заместителям цикла, но при этом подходят к молекуле изоксазолина с наиболее доступной стороны. Восстановление изоксазолинов водородом в присутствии амальгам происходит нестереоспецифично — по-видимому, из-за первоначального разрыва цикла по связи N—О с последующим восстановлением связи C—N. Такое же нестереоспецифическое раскрытие, приводящее к стерео-изомерной смеси аминоспиртов (3:2), наблюдается и при гидрировании над катализатором Адамса.
Раскрытие изоксазольного цикла происходит при каталитическом гидрировании, обычно используемые катализаторы — палладий на носителях, Ni-Ra, окись платины; выходы препаративные. В зависимости от условий восстановления образуются β-енаминокетоны XXX или р-дикетоны XXXII.
Синтез полифункциональных енаминокетонов LXXVI и β-дикетонов LXXVII осуществлен из изоксазолов LXXVIII путем последовательной модификации заместителей и восстановительного расщепления цикла в производных LXXIX; дикетоны получали кислотным гидролизом енаминокетонов.
Превращение изоксазолов через енаминокетоны в непредельные кетоны LXXXI и LXXXII путем расщепления по Берчу с последующим дезаминированием либо каталитическим гидрогенолизом цикла и последующим восстановлением ацилированного производного LXXXIII успешно применено в синтезе простагландинов. Использование изоксазольной стратегии для конструирования цепи простаноидов основано на синтезе изоксазолзамещенных циклопентанонов LXXX, расщепление гетероцикла в которых дает природную цепь простагландина.
Рециклизация енаминокетонов или их производных по карбонильной группе открывает возможности синтеза других азотсодержащих гетероциклов. Такой путь использован для разработки синтеза 8-азапростаноидов.
Аналогичным процессом рециклизации α-гидроксикетоенаминс LXXXVII из изоксазольных производных стероидов LXXXVIII является одностадийное каталитическое восстановление последних над Ni-Ra уксусной кислоте. В последние годы и основе этой реакции разработаны методы синтеза дигидрофуранонов LXXXIX, являющихся центральными структурными фрагментами целого ряда природных соединений.
Результаты исследований последних лет в достаточно полной мере отражают и достоинства, и проблемы изоксазольной стратегии синтеза функционализированных органических молекул. Выявлены различные структурные факторы стереоконтроля процессов образования, модификации и раскрытия изоксазольного цикла, найдены селективные реагенты, разработана методология выбора защитных групп при синтезе полифункциональных соединений, в ряде случаев удалось осуществить стереоселективный препаративный синтез. Для тех молекул, у которых удельный вес изоксазольного фрагмента существен, есть опыт по предсказанию стереонаправленности реакций, причем электронное и пространственное усложнение молекулы до определенного предела повышает стереоселективность всех этапов синтеза. Такую ситуацию мы как раз имеем при синтезе природных соединений, когда довольно большие объемы всей молекулы и заместителей гетероцикла являются факторами стереоконтроля, — до тех пор, однако, пока эти факторы не препятствуют возможности самой реакции. Стереохимические проблемы изоксазольного метода связаны в основном с разработкой новых селективных реагентов и техники селективных реакций. В настоящее время реализация задачи полного синтеза природных соединений и синтеза аналогов обеспечивает бурный прогресс в этой области циональных групп либо в цикл, либо в экзоциклическое положение; раскрытие цикла, приводящее к бифункциональному производному
1. Лахвич Ф.А., Е.В. Королева, А.А. Ахрем. Синтез, химические трансформации и проблемы применения производных изоксазола в полном химическом синтезе природных соединений // Химия гетероциклических соединений. 1989, №4, СС. 435-453
2. Петров А.А., Бальян Х.В., Трощенко А.Т. Органическая химия. – М.: Высш. шк., 1973. - 623 с.
3. Десенко С.М. Азагетероциклы на основе ароматических непредельных кетонов. Харьков: Фолио, 1998, 148с.
4. В. Ф. Травень. Органическая химия. Том 1. – М.: Академкнига, 2004, - 708 с.
5. Шабаров Ю.С. Органическая химия: В 2-х кн. - М.:Химия, 1994.- 848 с.
6. Джилкрист Т. Химия гетероциклических соединений. М.: Мир, 1996, 464с.
7. Терней А. Современная органическая химия: В 2 т. - М.: Мир, 1981. - Т.1 - 670 с; Т.2 - 615 с.
8. Лернер И.М.. Указатель препаративных синтезов органических соединений. Л.: Химия, 1982, 280с.
|
