|
1. Характеристика окремих електронів у молекулі і молекулярні електронні оболонки
2. Коливна структура електронно-коливного переходу. Принцип Френке-Кондора
3. Загальна характеристика обертової структури електронно-коливних смуг
4. Правила відбору і типи електронних переходів
1.
Характеристика окремих електронів у молекулі і молекулярні електронні оболонки
Розглянемо характеристики станів електронів в двохатомній молекулі (одноелектронних станів) і молекулярні електронні оболонки, в які групуються окремі електрони.
Спочатку розглянемо характеристики одноелектронних станів у молекулі незалежно від властивостей атомів, що її утворюють.
Важливою характеристикою стану окремого електрона в атомі є азимутальне квантове число l, що визначає значення орбітального моменту цього електрона. В залежності від значення l ми можемо розрізняти s-, p-, d-, f-електрони і одержувати оболонки, які заповнюються 2, 6, 10, 14 ... електронами.
В двохатомній або лінійній багатоатомній молекулі можна аналогічно наближено вважати, що окремий електрон рухається в полі ядер, що утворюють вісь молекули і інших електронів. В зв’язку з цим, основною наближеною характеристикою одноелектронних станів служить квантове число l, що визначає абсолютну величину проекції орбітального моменту електрона на вісь молекули. Так як величина цієї проекції рівна l2
= m2
, де ml
= 0; ±1; ±2 .., то l приймає значення l = |ml
| = 0, 1, 2, 3... . Енергія електрона в молекулі залежить лише від абсолютної величини проекції, так як на електрон в молекулі діє електричне поле.
Стани з l = 0, 1, 2, 3... по аналогії з l = 0, 1, 2, 3... для атомів позначають грецькими буквами:
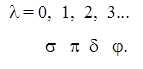
Згідно принципу Паулі, в молекулі не може бути двох електронів в одинакових квантових станах, відповідно, і з одинаковими наборами квантових чисел. Еквівалентні s-електрони (l = 0) можуть відрізнятися лише величиною ms
проекції спіна на вісь молекули, яке приймає значення ms
=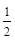 і ms
= – і ми одержимо молекулярну оболонку s2
, заповнену двома електронами. Еквівалентні p і d електрони можуть відрізнятися знаком проекції орбітального моменту і знаком проекції спіна на вісь молекули, тобто існують чотири різних стани: ml
= l, ms
=; ml
= l, ms
= –; ml
= –l, ms
=; ml
= –l, ms
= – і одержуються молекулярні оболонки p4
, d4
.., заповнені чотирма електронами кожна. Таким чином, в лінійних молекулах не може бути більше чотирьох еквівалентних електронів, тобто не існує молекулярних електронних оболонок, заповнених більше, ніж чотирма електронами. При цьому s-оболонки (l = 0) заповнюються двома електронами, а p- і d-оболонки (l > 0) – чотирма електронами. При інших однакових умовах енергія s-електронів менша за енергію p-електронів, а p- – менша за енергію d-електронів. Для лінійних молекул, що мають центр симетрії, одноелектронні стани можна, як і стани всієї молекули в цілому, розділити на парні g і непарні u. Таким чином, ми одержимо стани sg
, pg
, dg
, jg
… su
, pu
, du
, ju
і відповідно можемо говорити про парні і непарні електрони, що відповідає переходам з рівня v² = 0. Це пояснюється тим, що при кімнатній температурі майже всі молекули знаходяться на нижньому коливному рівні. Вигляд спектра випромінювання (люмінісценсії) залежить від характеру збудження. Якщо молекула збудження лише до деякого конкретного рівня v¢, то саме з нього і будуть здійснюватись переходи з випромінюванням, тобто виникає відповідна поперечна серія Деландра. і ms
= – і ми одержимо молекулярну оболонку s2
, заповнену двома електронами. Еквівалентні p і d електрони можуть відрізнятися знаком проекції орбітального моменту і знаком проекції спіна на вісь молекули, тобто існують чотири різних стани: ml
= l, ms
=; ml
= l, ms
= –; ml
= –l, ms
=; ml
= –l, ms
= – і одержуються молекулярні оболонки p4
, d4
.., заповнені чотирма електронами кожна. Таким чином, в лінійних молекулах не може бути більше чотирьох еквівалентних електронів, тобто не існує молекулярних електронних оболонок, заповнених більше, ніж чотирма електронами. При цьому s-оболонки (l = 0) заповнюються двома електронами, а p- і d-оболонки (l > 0) – чотирма електронами. При інших однакових умовах енергія s-електронів менша за енергію p-електронів, а p- – менша за енергію d-електронів. Для лінійних молекул, що мають центр симетрії, одноелектронні стани можна, як і стани всієї молекули в цілому, розділити на парні g і непарні u. Таким чином, ми одержимо стани sg
, pg
, dg
, jg
… su
, pu
, du
, ju
і відповідно можемо говорити про парні і непарні електрони, що відповідає переходам з рівня v² = 0. Це пояснюється тим, що при кімнатній температурі майже всі молекули знаходяться на нижньому коливному рівні. Вигляд спектра випромінювання (люмінісценсії) залежить від характеру збудження. Якщо молекула збудження лише до деякого конкретного рівня v¢, то саме з нього і будуть здійснюватись переходи з випромінюванням, тобто виникає відповідна поперечна серія Деландра.
Реклама

Загальний характер розподілу інтенсивностей в системі електронно-коливних смуг визначають, виходячи з принципу Франка-Кондона. Суть принципа полягає в слідуючому. Якщо параметри потенціальних кривих, між якими проходять електронно-коливні переходи, задані, то виникає питання, які з вказаних переходів будуть більш, а які менш імовірні. Відповідь на ці питання дає принцип Франка-Кондона, зміст якого полягає в слідуючому.
Молекула, на відміну від атома, складається з двох зв’язаних підсистем, рух яких проходить з суттєво різними швидкостями. Це сукупність електронів, яка є швидкою підсистемою, і сукупність ядер, яка є повільною підсистемою. При взаємодії молекули із світлом стан швидкої підсистеми змінюється надзвичайно швидко. В той же час коливання ядер проходить значно повільніше, зміна властивостей електронної оболонки при опроміненні здійснюється так швидко, що швидкості і положення ядер при цьому змінитися не встигають. Іншими словами, в процесі електронно-коливних переходів молекула виявляється у збудженому стані при тому ж значенні міжядерної віддалі, що і в основному стані. Звідси випливає, що на діаграмах кривих потенціальної енергії квантові переходи між різними електронними станами двохатомної молекули повинні зображуватись переважно вертикальними стрілками, які відповідають умові незмінності міжядерної віддалі в процесі переходу. Таким чином, найбільш імовірними будуть переходи, що відповідають збереженню віддалі між ядрами, тобто вертикальним лініям на графіку потенціальних кривих (мал. ). На мал. приведена схема коливних переходів випромінювання (а) і поглинання (б) в рамках електронного перехода.
Реклама

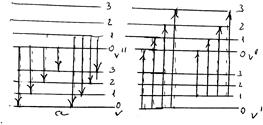
Мал. . Коливні переходи з випромінюванням (а) і поглинанням (б) в рамках одного електронного переходу.
Таку схему рівнів називають схемою Деландра. Серія ліній, що відповідає випромінюванню, називають поперечною серією Деландра, а поглинання – поздовжньою серією Деландра. Відповідно, в електронному спектрі поглинання або люмінісценсії повинно спостерігатись велика кількість смуг, що відповідають одному електронному переходу, але різним коливним переходам. Реальний спектр молекули виглядає простіше. В дійсності в поглинанні зазвичай є тільки одна поздовжня серія Деландра.
2. Коливна структура електронно-коливного переходу. Принцип Френке-Кондора
При переході між двома електронними станами, як правило, одночасно змінюються і коливна, і обертова енергія. Повна зміна енергії при переході
DЕ = DЕел
+ DЕкол
+ DЕоберт
,
Причому
DЕел
> DЕкол
> DЕоберт
.
Кожен електронний перехід між двома електронними термами (станами) характеризується певною коливною структурою, яка складається з сукупності смуг, а кожна смуга характеризується певною обертовою структурою, яка предятавляє собою сукупність окремих ліній.
Розглянемо спочатку коливну структуру, яка, на противагу обертовій, не залежить беспосередньо від властивостей симетрії комбінуючих електронних станів і визначається видом кривих потенціальної енергії для цих станів. Положення смуг коливної структури визначається різницею DЕкол
= DЕ¢кол
– DЕ²кол
енергії коливань DЕ¢кол
для верхнього електронного рівня і енергії коливань DЕ²кол
для нижього електронного рівня, які, якщо обмежитися двохчленною формулою, рівні:
DЕ¢кол
= n¢е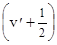 – х¢е
n¢е
– х¢е
n¢е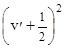 ;
;
DЕ²кол
= n²е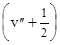 – х²е
n²е
– х²е
n²е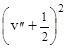 .
.
Значення постійних nе
і хе
, тобто частот коливань і коефіцієнтів, які визначаються ангармонічністю, для різних електронних станів в загальному різні. Як правило, для збудженого стану міцність зв’язку менша, а рівноважна віддаль між ядрами більша, ніж для основного стану, і крива потенціальної енергії проходить більш полого. В результаті частота коливань nе
у збудженому стані менша, ніж у нормальному, а із збільшенням збудження вона зменшується. Тому n²е
> n¢е
відповідно і віддалі між коливними рівнями для верхніх електронних станів менші, ніж для нижніх станів. Наприклад, для основного електронного стану 1
S+
молекули СО n²е
= 2169 см–1
, а для збудженого стану n¢е
= 1515 см–1
.
Частота nе
беспосередньо зв’язана з силовою постійною (K) молекули в даному електронному стані. Зменшення nе
при збудженні молекули означає зменшення силової постійної, яка проходить, як правило, одночасно із зменшенням міцності зв’язку, тобто енергії дисоціації.
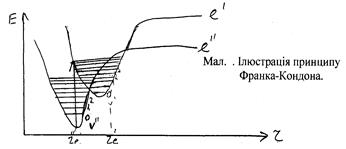
Вертикаль проводиться з центра рівня v² = 0, тобто з мінімума потенціальної кривої, так як для v² = 0 найбільш імовірне утворення молекули відповідає r = re
.
Можливі три основні випадки розподілу інтенсивностей в серії Деландре v² = 0:
1) Криві верхнього і нижнього станів точно розміщуються одна над одною. В цьому випадку принцип Франка-Кондона виконується для переходу v² = 0 ® n¢ = 0. Для більш високих n¢ кінетична енергія і швидкість коливання молекул рівні нулю, як для нульового рівня, лише в точках повороту А і В. За точками повороту прийняті точки, що відповідають найбільшим відхиленням атомів від положення рівноваги при коливанні. Для переходу v² = 0 ® n¢ = 1 є незначна зміна положення атомів і тому ця смуга у спектрі присутня, але з невеликою інтенсивністю. Подальші смуги ще менш інтенсивні.
2) Потенціальна крива збудженого стану дещо зміщена вправо. В цьому випадку найбільш імовірним стає перехід v² = 0 ® n¢ = 1.
3) Верхня потенціальна крива ще більше зміщена вправо. Це відповідає суцільному спектру дисоціації молекули. В результаті такого переходу молекула дисоціює.
Для серій смуг випромінювання v¢ ¹ 0 (так же як і для серій смуг поглинання з v² ¹ 0) розподіл інтенсивностей буде відрізнятись від розглянутого. Дійсно, для молекули, що знаходиться на коливному рівні з v¢ ¹ 0, найбільш імовірними віддалями між ядрами будуть віддалі, що відповідають точкам поворота А і В (мал. ). Саме з цих точок і будуються вертикалі Франка-Кондона.
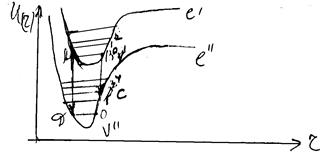
Мал. Використання принципу Франка-Кондона для електронно-коливних переходів з рівнів v¢ ¹ 0.
Перехід з точки В приведе в точку С нижнього стану. Перехід з точки А приведе відповідно в точку D. Таким чином, одержуємо два значення v², для яких імовірність переходу з даного v¢ максимальна. Відповідно, слід чекати появи двох максимально інтенсивних смуг в серії v¢ = 1.
Якщо уявити розміщення смуг з максимальною інтенсивністю (максимально імовірні переходи) в таблиці всіх можливих коливних переходів (таблиця Деландра), то вони розмістяться по параболі, яка називається параболою Кондона (мал. ). Тільки параболи розходяться тим ширше, чим більше зміщення верхньої потенціальної кривої відносно нижньої. Парабола Кондона буде тим вужча, чим менше зміщені одна відносно одної потенціальні криві комбінуючих електронних станів. В граничному випадку, коли вони лежать одна над одною, парабола вироджується в пряму.
| n²
|
0
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
| n¢
|
| 0
|
*
|
|
|
|
|
|
|
|
| 1
|
*
|
|
|
*
|
|
|
|
|
| 2
|
|
*
|
|
|
|
*
|
|
|
| 3
|
|
|
*
|
|
|
|
|
*
|
| 4
|
|
|
|
*
|
|
|
|
|
Мал. Парабола Кондона: * – відмічені найбільш імовірні переходи
3.
Загальна характеристика обертової структури електронно-коливних смуг
Обертова структура електронно-коливних смуг визначається зміною обертової енергії при відповідному переході DЕоберт
= DЕ¢оберт
+ DЕ²оберт
. Для обертових рівнів верхнього електронно-коливного стану маємо: DЕ¢оберт
= 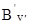 hy¢(y¢ + 1) і для обертових рівнів нижнього електронно-коливного стану маємо аналогічно: DЕ²оберт
= hy¢(y¢ + 1) і для обертових рівнів нижнього електронно-коливного стану маємо аналогічно: DЕ²оберт
= 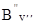 hy²(y² + 1). Таким чином, без врахування поправок на центробіжне розтягування молекули одержимо: DЕоберт
= = hy¢(y¢ + 1) – hy²(y² + 1). Таким чином, без врахування поправок на центробіжне розтягування молекули одержимо: DЕоберт
= = hy¢(y¢ + 1) – 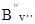 hy²(y² + 1). Обертові постійні для верхнього і нижнього стану різні і лише в деяких випадках приблизно співпадають. Їх основна відмінність визначається зміною рівноважної віддалі rе
між ядрами при переході молекули з одного електронного стану в інший. Як уже вказувалося, при електронному збудженні молекули Еел
збільшується; це приводить до збільшення моменту інерції і до зменшення обертової постійної. Тому В¢ < В². hy²(y² + 1). Обертові постійні для верхнього і нижнього стану різні і лише в деяких випадках приблизно співпадають. Їх основна відмінність визначається зміною рівноважної віддалі rе
між ядрами при переході молекули з одного електронного стану в інший. Як уже вказувалося, при електронному збудженні молекули Еел
збільшується; це приводить до збільшення моменту інерції і до зменшення обертової постійної. Тому В¢ < В².
При електронних переходах в загальному випадку для обертового квантового числа у має місце звичайне правило відбору для моментів кількості руху: Dу = 0; ±1. Для зміни обертової енергії при електронно-коливних переходах можна використати всі формули, виведені для зміни обертової енергії при чисто коливних переходах. Ми маємо R-, P- і Q-гілки:
nR
= n0
+ 2B¢v
+ (3B¢v
– B²v
) j + (3B¢v
– B²v
) j2
;
nP
= n0
– (2B¢v
+ B²v
) j + (B¢v
– B²v
) j2
;
nQ
= n0
+ (B¢v
– B²v
) j + (B¢v
– B²v
) j2
.
Відповідні R-, P- і Q-переходи показані на малюнку .
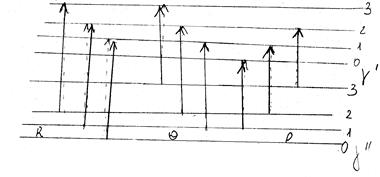
Мал. . R-, P- і Q-гілки обертових переходів в електронно-коливних спектрах Dj = +1(R);
Dj = +1(R); Dj = 1(Q); Dj = –1(P).
Якщо ввести к формули ціле число m, де
m = j¢ для R-гілки m = 1, 2, 3..,
m = –j² для P-гілки m = –1, –2, –3..,
m = j¢ = j² для Q-гілки m = 1, 2, 3..,
то формули приймуть вигляд:
R- і P-гілки: DЕ = n0
+ (В¢ + В²) m + (В¢ – В²) m2
;
Q-гілка: DЕ = n0
+ (В¢ – В²) m (m + 1).
Тут n0
– частота електронно-коливного переходу, що відповідає m = 0.
4. Правила відбору і типи електронних переходів
Основні типи електронних переходів в двохатомних молекулах визначаються правилами відбору, як загальними, так і більш окремими. Перш за все, для повного моменту кількості руху молекули справедливе загальне правило відбору Dj = 0, ±1 (з додатково забороненим переходом j¢ = 0 ® j² = 0.
У відповідності з поділом станів молекули на додатні і від’ємні є правило відбору, що дозволяє переходи між станами одинакової симетрії + « –. Для молекул, що мають центр симетрії, парні електронні стани комбінують з непарними g « u. Важливою характеристикою електронних станів молекули є квантове число L, що визначає величину проекції орбітального моменту кількості руху на вісь молекули і рівне абсолютному значенню цієї проекції. Для L має місце правило відбору: DL = 0, ±1. Таким чином, можливі переходи S–S, S–P, P–P, P–D, D–D. Ці переходи можна розділити на три типи:
1 тип – переходи S–S, DL = 0, L = 0;
2 тип – переходи S–P, P–D, DL = 1;
3 тип – переходи P–P, D–D, DL = 0 і DL ¹ 0.
Відмітимо, що при позначенні електронних переходів спочатку записують символ верхнього стану, а потім нижнього і, відповідно, перехід записується L¢–L². Наприклад, S–P означає перехід між верхнім станом S і нижнім P-станом.
Для переходів першого типу є додаткове обмеження, зв’язане з тим, що S-стани можуть бути додатніми (S+
) і від’ємними (S–
). Дозволені переходи лише між S-станами одної симетрії: S+
–S+
; S–
–S–
.
Переходи трьох типів відрізняються своєю обертовою структурою. Для переходів першого типу S–S є додаткове правило відбору, яке забороняє Q-гілку і виникають тільки R- і Р-гілки. Для переходів другого типу, зокрема S–P і P–S проявляються всі три гілки R, P і Q. Для переходів третього типу, зокрема P–P, також можливі всі три гілки, але в Q-гілці інтенсивність різко зменшується із збільшенням квантового числа і ця гілка слаба.
При участі спіна одержується подальша класифікація типів електронних переходів. Як і для атомів, є наближене правило відбору для повного спінового механічного моменту S, DS = 0. Найбільш простою є обертова структура переходів 1
S±
– 1
S±
. Для них одержуються R- і Р-гілки (Q-гілка відсутня).
Суцільні спектри поглинання і випромінювання двохатомної молекули. Поряд з дискретними спектрами для лінійних молекул можуть виникати і суцільні спектри. Вони утворюються при переходах між двома станами, по крайній мірі один з яких відповідає неперервній послідовності значень енергії.
Суцільні спектри можуть одержуватися при переходах між дискретними нижніми рівнями і неперервними верхніми рівнями, між неперервними нижніми і дискретними верхніми. Суцільні спектри також будуть виникати при переходах між неперервними нижніми і неперервними верхніми рівнями. Розподіл інтенсивностей у суцільних спектрах, як і в дискретних, буде визначатися принципом Франка-Кондона, тобто найбільш імовірними будуть переходи, які зображаються вертикальними лініями, що з’єднують потенціальні криві комбінуючих рівнів. Правила відбору в цьому випадку такі, як і для переходів між різними електронними станами.
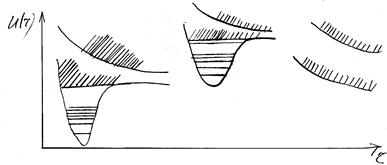
Мал. Схема утворення неперервних спектрів для стійких і нестійких спінів.
Визначення з досліду положення nгран.
– межі сходження смуги, тобто межі дисоціації має суттєве значення. По положенню цієї межі можна визначити з досить високою точністю енергію дисоціації. Якщо через n0
позначити енергію електронного збудження і через D¢ – енергію дисоціації збудженого електронного стану, то nгран
= n0
+ D¢ і знаючи n0
можна знайти D¢.
|
