Лигандообменная хроматография основана на образовании координационных связей между сорбентом и разделяемыми ионами или молекулами. Лигандообменная хроматография применима только для разделения соединений, содержащих донорные гетероатомы или кратные связи. Ионы переходных металлов, находящиеся в неподвижной фазе, являются акцепторами электронов и легко вступают в координационное взаимодействие с электронодонорными атомами функциональных групп разделяемого соединения. Для проведения лигандного разделения необходимо наличие склонных к координации органических соединений и комплексообразующего катиона металла. Такое разделение характеризуется обратимостью процесса и высокой скоростью обмена лигандов. Лигандный обмен применяют в жидкостной колоночной, тонкослойной и газовой хроматографии, но наибольшие успехи были достигнуты в ВЭЖХ.
Лигандообменную хроматографию применяют для разделения в водной среде соединений, представляющих большой интерес для органической химии и биохимии: аминов, аминокислот, белков, нуклеотидов, пептидов, углеводов. При этом в вчестве комплексообразующих используют ионы меди, цинка, кадмия, никеля, серебра и железа. Ионы ртути и серебра в неполярной среде алифатических углеводородов образуют лабильные комплексы с ненасыщенными и ароматическими углеводородами. Большими достоинствами лигандообменной хроматографии является ее селективность и отсутствие жестких требований к сорбенту, который может быть прочно связан ионами металла или только пропитан солями металла.
Аминокислоты занимают две координационные позиции, т.e. являются бидентатными лигандами, что резко повышает прочность их связывания за счет образования хелатного комплекса.
Лигандообменная хроматография оптически активных соединений основана на образовании лабильных координационных соединений, в которых с центральным ионом металла-комлексообразователя одновременно координирована молекула расщепляющего асимметрического реагента и один из подлежащих разделению энантиомеров. Для осуществления лигандообменной хроматографии необходимо наличие в расщепляющих реагентах и в разделяемых лигандах донорных гетероатомов серы, кислорода, азота, способных координироваться с ионом металла. Наблюдается хорошая координация α-аминокислот и ионов меди, цинка и никеля. Донорные атомы образуют плотно упакованную координационную сферу вокруг центрального иона металла, при этом разделяемые лиганды вступают в тесный контакт с расщепляющим реагентом. Этим и объясняется высокая эффективность распознавания реагента, т.e. энантиоселективность.
Такой принцип расщепления рацематов–лигандообменная хроматография – впервые был применен с использованием полистирольного сорбента, ковалентно связанного с остатками оптически активной природной аминокислоты L-пролина. Сорбент прочно координировал двухвалентную медь, оставляя в ее основной координационной плоскости две вакантные позиции для связывания подвижного лиганда молекулы L- или D-аминокислоты. Оказалось, что остаток L-пролина проявляет настолько высокое сродство к D-изомерам аминокислот, что последние пришлось даже вымывать из колонки раствором аммиака, который, координируясь с ионами меди, вытеснял подвижный лиганд из сорбционного комплекса, в то время как L-изомеры десорбировались водой.
Реклама

Взаимодействием аминогруппы оптически активной аминокислоты с хлорметиль-ной группой полистирола в присутствии иодида натрия синтезировано более 50 сорбентов, имеющих асимметрические атомы. Оказалось, что циклические аминокислоты пролин и оксипролин обладают максимальной энантиоселективностью и количественно расщепляют рацематы практически всех аминокислот, а также многие оксикислоты,
диамины, аминоспирты. Достоинством полистирольных асимметрических сорбентов являются высокая химическая стабильность, большая обменная емкость и возможность препаративного расщепления рацематов. Иногда за один цикл на 300 г. сорбента удается расщепить до 20 г. рацемата. Появились лиган-дообменники с привитыми к сили-кагелю аминопропильным или полученным из него дитиокарбаматным радикалом.
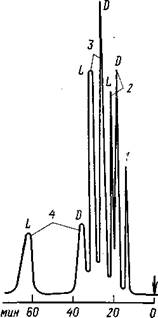
Рис. 1. Хроматограмма рацематов аминокислот, полученная на колонке размером 100Х1 мм (стекло) с асимметрическим полистирольным анионитом (7,5 мкм), подвижная фаза – 0,25 М ацетат натрия и 1,5Х10-3
М ацетат меди, рН=5,2, детектор–УФ (260 нм): 1 – лизин; 2 – аланин; 3 – серии; 4 – лейцин
Лигандообменную хроматографию с успехом применяют для производства оптически чистых меченных тритием аминокислот.
Весьма важное применение лигандообменной хроматографии – быстрое разделение асимметрических энантиомеров без предварительного отделения от сопутствующих примесей. В лигандообменной жидкостной хроматографии в отличие от лигандообменной газовой хроматографии не требуется предварительное превращение энантиомеров в легколетучие соединения. Обычно в качестве сорбента применяют полистирол, к которому привит радикал оптически активного бензилзамещенного пропилендиамина.
Для сокращения времени анализа, повышения его чувствительности и точности требуется повышение эффективности колонки, что достигается за счет уменьшения частиц полистирольного сорбента. Перспективным является получение сорбентов для лигандообменной хроматографии на основе пористых силикагелей, химически модифицированных активными лигандами. Поверхность силикагеля связывается через n-пропиленовые мостики с L-оксипролиновыми лигандами.
В настоящее время создано и испытано около десятка асимметрических силикагелевых сорбентов. Хотя в большинстве сорбентов использованы оптические изомеры пролина и оксипролина, они различаются сродством к различным аминокислотам и порядком их элюирования; это связано с тем, что подвижный лиганд фиксируется в комплексе не только координацией карбоксильной и аминогруппой с ионом меди, но и взаимодействием бокового радикала аминокислоты с ближайшим окружением координационного центра, что определяет в конечном итоге порядок удерживания антиподов аминокислот сорбенте.
Реклама

Поскольку сорбенты для лигандообменной хроматографии выпускаются, предложены некоторые модификации метода, позволяющие использовать традиционные сорбенты. Так, N-алкилированные оптически активные аминокислоты сорбируются на обращенно-фазном силикагеле. В алкильном радикале должно быть больше 5–6 углеродных атомов. В этом случае модификатор прочно удерживается на сорбенте и не смывается водным элюентом, в который добавляют следовые количества меди.
Определение микропримесей
ВЭЖХ является надежным методом анализа микропримесей–веществ с концентрацией 0,01%.
В жидкостной хроматографии применяют селективные детекторы (амперометрический, флуориметрический и др.), способные детектировать очень малое количество вещества. Очистка образца до ввода в жидкостной хроматограф минимальна, нередко его вводят без предварительной обработки, и без получения производных, что часто невозможно при применении других методов анализа. Наконец, в жидкостной хроматографии возможно создание уникального диапазона селективных взаимодействий за счет изменения подвижной фазы, что значительно улучшает разрешающую способность всей хроматографической системы. Работа с микропримесями налагает ряд требований на весь процесс разделения. Особенное значение имеет разрешающая способность колонки, выбор детектора, предварительная обработка образца и построение калибровочного графика. Правильный выбор условий хроматографирования позволяет повысить чувствительность, надежность и воспроизводимость результатов, что очень актуально при работе с микропримесями.
Предварительная подготовка образца и его ввод в хроматограф
Перед анализом микропримесей для повышения чувствительности желательно провести концентрирование соединения методами, описанными в соответствующем разделе. Предпочтительно применять оборудование, позволяющее проводить автоматическую предварительную обработку образца, в том числе экстрагирование, измельчение, фильтрацию и автоматический ввод в хроматограф.
Для получения высокой чувствительности нужно вводить максимально возможный объем образца. При работе в изократическом режиме на высокоэффективных аналитических колонках оптимальный объем образца составляет от 100 до 500 мкл. Ввод большого объема разбавленного образца позволяет компенсировать относительно низкую чувствительность некоторых детекторов для жидкостной хроматографии.
При небольших количествах образца анализ проводят на колонках с внутренним диаметром 1–2 мм, что уменьшает его расход. Используя в качестве растворителя пробы слабый растворитель, можно наносить на колонку большие объемы образца. При этом он накапливается на входе в колонку, так как к' для компонентов образца велико и в колонке происходит концентрирование микропримесей, что значительно повышает чувствительность обнаружения. Пользуясь этим приемом, можно обнаружить некоторые малополярные органические соединения, например ароматические углеводороды, присутствующие в виде микропримесей в сточных водах, на колонке с обращенной фазой.
Для обеспечения наибольшего концентрирования растворитель пробы должен быть по возможности слабым, например, содержать 90–95% воды. В дальнейшем для элюирования пробы силу подвижной фазы резко увеличивают. Концентрирование на колонке позволяет в некоторых случаях вводить 1 л и более образца при конечном объеме зоны интересующего нас вещества менее 50 мкл (градиентное элюирование), т.е. концентрировать вещество в 20 000 раз. Можно повысить чувствительность обнаружения, проводя концентрирование образца и вне колонки, например, выпаривая растворитель. Однако надо быть уверенным, что при этом не происходит потеря или видоизменение примесей. Кроме того, при таком процессе значительно повысится содержание основных компонентов в образце, что приведет к перегрузке по ним колонки и затруднит анализ. Вещества, удерживаемые сильнее, чем примеси, могут быть после анализа, удалены из колонки не только за счет повышения силы растворителя, но и путем обратной промывки колонки.
Разрешающая способность колонки
Иногда удается провести разделение на обращенной фазе таким образом, что основные компоненты образца элюируются со временем, близким к t0, а микропримеси удерживаются на колонке, что облегчает их количественное определение. Оценка содержания микропримеси наиболее надежна, когда ее пик регистрируется до пика преобладающего в образце компонента. Элюирование пика микропримеси на хвосте преобладающего компонента сильно ухудшает ее количественное определение. Любое хроматографическое разделение на колонке приводит к разбавлению образца. При анализе микропримесей возникает проблема разделения при минимальном разбавлении. Степень разбавления введенного образца может быть выражена уравнением
С
макс/С
0 = VsN0,5
/[Vr(2π)0,5
],
где С
макс – Концентрация, соответствующая максимуму пика; С0 – исходная концентрация в образце;
Vs
– вводимый объем;
N
– число теоретических тарелок для данной колонки;
Vr
– удерживаемый объем микропримеси.
Из уравнения видно, что снижения степени разбавления. а значит, увеличения чувствительности определения микропримеси можно достигнуть за счет увеличения вводимого объема, ^использования колонки с большим числом теоретических тарелок или за счет снижения объема удерживания микропримеси. К увеличению N ведет применение эффективных колонок, заполненных частицами с размером менее 10 мкм. При анализе микропримесей желательно применение коротких колонок (5–10 см) с размером частиц 3–5 мкм.
Большое влияние при анализе микропримесей оказывает изменение селективности системы и повышение коэффициента разделения а
, что достигается изменением состава подвижной фазы и выбором оптимального хроматографического режима. Кроме того, как видно из уравнения
C
макc/С
0 =(α–1)/α
изменение селективности может значительно снизить степень разбавления пика.
Детекторы
При анализе микропримесей работают с селективными детекторами, чувствительными к интересующему веществу. Предельная чувствительность детектора зависит от отношения сигнала к шуму самого детектора и от его способности реагировать на микропримеси в образце. Перед выполнением анализа необходимо эти микропримеси идентифицировать. Наиболее часто при анализе микропримесей применяют высокочувствительные фотометрические детекторы, работающие в УФ-области, и спектрофотометры. УФ-фотометры, имеющие чувствительность более 0,002 е.о.п. на всю шкалу при шуме 1%, позволяют обнаруживать нанограммовые количества веществ, умеренно поглощающих в УФ-области, а флуориметры – даже пикограммовые. Для достижения максимальной чувствительности желательно работать при длине волны, соответствующей максимуму поглощения в УФ-спектре вещества. Получение соответствующих производных после колонки повышает селективность анализа и позволяет проводить его при максимальной предварительной очистке образца.
Примеси, способные к окислению или восстановлению в электролите, наиболее целесообразно определять чувствительными электрохимическими детекторами.
Калибровочные графики
При количественном анализе микропримесей не так важна высокая точность измерений, как их надежность, что достигается получением зон, свободных от посторонних соединений. Так как точность около 10% считается вполне удовлетворительной, пики можно оценивать по их высоте, меньше зависящей от четкости отделения микропримеси от соседних зон, пики которых могут накладываться на пики анализируемых микропримесей. Хотя оценка по площадям пиков более точна, калибровка по высоте пиков удобнее.
В ВЭЖХ калибровочные графики микропримесей обычно линейны и проходят через начало координат. Чтобы избежать возможных частичных потерь образца, проводят калибровку методом внутреннего стандарта (по соотношению высот пиков), причем внутренний стандарт известной концентрации добавляют до предварительной обработки образца. В этом случае потери учитывают за счет эквивалентного изменения концентрации внутреннего стандарта и образца. Стандартный образец, близкий по свойствам микропримеси, добавляют, когда трудно выделить анализируемое вещество в чистом виде, чтобы построить калибровочный график. Подробнее калибровка по методу внутреннего стандарта рассмотрена в гл. 10.
При анализе сложных смесей, в частности в тех случаях, когда наблюдаются эффекты влияния матрицы или когда получение в чистом виде исходного образца, для которого строится калибровочный график, затруднительно, применяют метод добавления стандарта. Наличие других компонентов в образце может влиять на степень удерживания и (или) высоту пика интересующего вещества. Метод добавления стандарта состоит в следующем. Образец разделяют в хроматографе и измеряют высоту hx
интересующего нас пика. После этого добавляют известное весовое количество Wx
вещества Х и повторяют разделение, определяя высоту пика h'x
вещества X. Для вещества Х строят калибровочный график и, определив калибровочный коэффициент Sx=(h'x
–hx
)/Wx
, вычисляют количество вещества Х в исходном образце как hx
/Sx
. Для проверки линейности графика берут несколько значений Wx
. Метод добавления стандарта не обеспечивает поправки на нестабильность базовой линии, на помехи со стороны других компонентов и т.д. Эти помехи должны быть выявлены до добавления стандарта.
Методом, альтернативным методу добавления стандарта, является получение образца без анализируемого вещества и использование этого образца как матрицы для подготовки стандартов при обычной калибровке.
При определении микропримесей нужно придерживаться следующих рекомендаций.
1. Проводить предварительное концентрирование образца, а в ряде случаев и предварительную его очистку.
2. Использовать большие объемы образца и короткие эффективные колонки, заполненные частицами сорбента менее 10 мкм, особенно микроколонки.
3. Применять наиболее чувствительные детекторы с высоким отношением сигнала к шуму.
4. Применять хроматографические системы, обеспечивающие высокую разрешающую способность для микропримесей (α>1,2) при относительно низких значениях к' (0,5–1,5).
Если это возможно, то разделение следует проводить в обращенно-фазном варианте, когда основной компонент выходит в зоне to
. 5. Калибровку проводить по высоте пиков.
Литература
1. Kucera P./J. Chromatogr, 1980, v. 198, p. 93–109.
2. Bowermaster J., McNair Я./J. Chromatogr, 1983, v. 279, p. 431–438.
3. Scott R.P.F./Adv. Chromatogr., 1983, v. 22, p. 247–294.
4. Van der Berg J.H.M., Horsels H.W.M., Groenen R.J. Af./Chromatographia, 1984, v. 18, No. 10, p. 574–578.
5. Freebairn K.W., Knox J. Я./Chromafographia, 1984, v. 19, No. 1, p. 37–47.
6. Schoenmakers P. /, Billiet H.A.H., DeGalan L./J. Chromatogr, 1981, v. 205, p. 13–30.
7. Jandera P., Churacek J./J. Chromatogr, 1980, v. 192, No. 1, p. 19–36.
8. Lawrence J.F., Frel R.W. Chemical Derivatisation in Liquid Chromatography. Amsterdam, Elsevier, 1976. 277 p.
9. Morris C. /., Morris P. Separation Methods in Biochemistry. N. Y, J. Wiley, 1976. 267 p.
10.Blau K., King G.S. Handbook of Derivatives for Chromatography London, Heyden, 1977. 784 p.
11.Kissinger P.T. e. a./J. Chromatogr. Sci, 1979, v. 17, p. 137.
12.Lawrence J.F. Organic Trace Analysis by Liquid Chrcpiafography. N.Y., Acad. Press, 1981. 288 p.
13.Roos R.W./J. Chromatogr. Sci, 1976, v. 14, p. 505.
14.Nachtmann P., Budna K.W./J. Chromatogr, 1977, v. 136, p. 279.
15.Clark C.R., Wells M.M./J. Chromatogr. Sci, 1978, v. 16, p. 332.
|
