|
Тема:
Методи аналізу рідких, твердих і газоподібних речовин
Рівне - 2010
Урок № 79-80 тема: Проведення простих видів аналізу за прийнятою методикою без попереднього поділу компонентів. Метод визначення вологи за Дином-Старка, заснований на відгонці води від аналізуючої речовини (при визначенні вмісту вологи в нафтопродуктах). Методи визначення густини рідин за допомогою ареометрів і ваг Мора-Вестфаля. Набір ареометрів для різних інтервалів густини. Метод гідравлічного зважування на вагах Мора-Вестфаля. Визначення густини з точністю до третього десяткового знака.
Визначення вологи
Наявність вологи в органічних сполуках проводять за допомогою проби на потріскування. Органічну сполуку нагрівають, якщо чути потріскування то це говорить про те, що в маслі, спирті, паливі міститься волога, якщо речовина тверда то вологу визначають методом висушування до постійної маси в сушильні шафі при температурі на 10˚-15˚С вище за температуру плавлення. Наважку із речовини 5-10 г взятої з точністю до 0,0001 г поміщають у висушений і зважений бюкс. Бюкс із речовиною висушують до постійної маси, вміст вологи обчислюють за формулою:
x = g – g1/g * 100
Вологість речовин, які розкладаються при нагріванні визначають за методом Діна і Старка. Цей метод оснований на відгонці води з не розчинному воді органічними рідинами, які утворюють з водою азотропні суміші, як розчинник використовують Бензин марки Галоша, перед тим його зневоднюють і фільтрують. Для зневоднення розчинник збовтують сульфатом натрію, суміш відстоюється, а потім фільтрують. В колбу поміщають 100 г нафтопродукту, зважують на технічних вагах, додають 100 мл зневодненого розчинника, з’єднують з холодильником, нагрівають пальником, утворюються пари, які конденсуються в холодильнику і стікають в логушку, вода збирається в її нижній частині. Перегонку проводять до тих пір, поки рівень води в логушці не буде змінюватись протягом години. Вміст вологи обчислюють за формулою:
W = V/g * 100%
де V – об’єм води в приймачі лопушки;
g – наважка продукту.
Визначення густини
Густина – це відношення маси речовини до її об’єму (г/мл), за одиницю приймають масу 1 см води, при температурі 4˚С. Густина залежить від температури, чим вища температура, тим менша густина. Якщо густину визначають при інших температурах, то перераховують густину при цій температурі ( , Y - поправка). Густину можна визначити аерометричним методом і пікнометричним, методом гідростатичного зважування на вагах Вестфаля-Мора.
Реклама

Для визначення густини використовуючи ареометри,
які являють собою скляну циліндричну посудину, верхня частина, якої закінчується трубкою, а нижня має кульку, яка містить баласт, який заставляє ареометр плавати вертикально. На трубці ареометра нанесені поділки з позначенням густини рідини. Для більшої точності використовують набір ареометрів, шкали яких охоплюють різний діапазон густин. Ареометр поміщаємо в посудину з рідиною, щоб він не торкався стінок циліндра, очікують 2-3 хв для бензину, а для в’язких продуктів до 15 хв і знімають покази на ареометрі по верхньому краю меніска. Вимірювання проводять декілька разів, поки розходження між двома параметрами визначень не перевищує 0,04
Визначення густини пікнометричним методом.
Пікнометр являє собою скляну посудину з міткою об’ємами від1 до 100 мл. Пікнометр промивають хромовою сумішшю, дистильованою водою, спиртом, висушують у сушильній шафі при температурі 100˚С, охолоджують в ексикаторі і зважують до 0,0002 г до постійної маси, а потім його калібрують. Для цього наповнюють дистильованою водою витримують термостат при 20˚С, доводять воду на шийці до мітки за допомогою полосок фільтрувального паперу, закривають кришкою і зважують, із 6-7 результатів беруть такий, які відрізняються на 0,005 г. Знаходять об’єм пікнометра:
V = g2 – g1/0.99823
g1 – маса пустого пікнометра;
g2 – маса пікнометра з водою;
0.99823 – маса 1 см води при температурі 20˚С.
Воду виливають, сполоскують спиртом, висушують у сушильній шафі при температурі 100˚С, охолоджують, наливають аналізуючи рідину вище мітки, поміщають в термостат при температурі 20˚С, витримують 15-20 хв за допомогою полосок фільтрувального паперу доводимо до мітки, зважуємо і знаходимо густину при температурі 20˚С:
= g2 – g1/V
V - об’єм пікнометра.
Для темних продуктів рівень рідини в пікнометрі встановлюють по верхньому меніску, а для світлих по нижньому.
Визначення густини на вагах Вестфаля-Мора.
Ваги мають коромисло, скляний поплавок, який поміщають в циліндр з аналізуючою речовиною, на одному плечі коромисла є противага і стрілка, яка в момент рівноваги встановлюється вертикально, чи на нуль. На другому кінці нанесено 10 поділок на, які навішуються різноважки. Поміщають аналізуючий розчин і добиваються за допомогою різноважок нуля, знімають покази густини.
Реклама

Урок № 81-82 тема:
Метод визначення температури крапле падіння. Необхідна апаратура (термометри, мішалки, пробірки, склянки, секундомір тощо), реактиви (олива вазелінова і медична, гліцерин, лід). Методи визначення температури плавлення горючих матеріалів. Прийоми введення проби в капіляр, її ущільнення. Визначення температури початку і кінця плавлення (момент перетворення всієї проби в рідину). Метод визначення температури застигання горючих матеріалів. Підготовка проби речовини і приладу – важлива умова правильного виконання аналізу. Методи визначення температури спалаху в приладах відкритого типу (у вигляді тиглів) і температури спалаху в приладі Мартене-Пенського. Устрій і призначення основних частин приладу Мартене-Пенського повітряної ванни із сорочкою, кришки з заслінкою, запальної лампочки, мішалки, термометра.
Визначення температури плавлення, крапле падіння
Тверді органічні речовини розплавляються в деякому інтервалі температур. З підвищенням температури вони розм’якшуються тому, за температуру плавлення нафтопродукту приймають температуру при, якій утворюється рідка краплина і називається температурою крапле падіння
. Отже, те крапле падіння показує температуру при, якій нафтопродукт переходить у рідкий стан. Температуру плавлення визначають за методом Жукова, для цього аналізуючий продукт розплавляють і нагрівають на 10˚-20˚С вище ніж очікуваної температури плавлення. Наливають на ¾ висоти підігрітий прилад Жукова, вставляють термометр так, щоб ртуть знаходилась на половині висоти досліджуваного продукту. Прилад розплавлення продукту залишається у спокої до тих пір поки температура не буде перевищувати очікуваної температури плавлення продукту на 3˚-4˚С, далі вміст приладу інтенсивно струшують, продукт починає мутніти і пінитися, після цього прилад ставлять на стіл і по секундоміру через 10-15 хв знімають покази термометра до повного затвердіння продукту.
За температуру плавлення приймають температуру на, якій стовпчик ртуті термометра затримувався найдовше.
Визначення температури спалаху і самозаймання
Температура спалаху є температура при, які пари речовини нагрітої в певних умовах, утворюють з навколишнім повітрям суміш, що спалахує при піднесені до неї полум’я. Якщо нафтопродукт нагріти вище температури спалаху, то наступає такий момент коли при піднесені полум’я він загоряється, температура при якій продукт загоряється і горить не менше 5 хв називається температурою самозаймання
.
Температуру спалаху і самозаймання характеризує ступінь вогненебезпечності продукту, наявність вологи, вміст легковипаровувальних речовин. За температурою спалаху можна легко виявити на наявність домішок, від температури спалаху залежить можливість використання масел в механізмах з нагріваючими поверхнями. Температуру спалаху можна вимірювати на апаратах Бренкліна з відкритим тиглем діаметром65 мм, металічного штативу, піщаної бані, термометра і газового пальника. Перед виконанням аналізу тигль промивають бензином, висушують і підігрівають на полум’ї пальника, ставлять у піщану баню так, щоб між дном тигля і дном бані був шар піску товщиною 5-8 мм, рівень піску повинен бути на висоті 12 мм від верхнього краю тигля. Тигль поміщають зневоднений і охолоджений нафтопродукт. Проводять нагрівання піщаної бані, спочатку 10˚С за 1 хв, а за 40˚С до очікуваної температури спалаху швидкість нагрівання зменшують до 4˚С за 1 хв, за 10˚С до очікуваної температури спалаху починають дослідження, через кожні 2˚С підносять тліючу скибочку, або запалювальну трубочку, за момент спалаху приймається поява на тлуопродуктом голубуватого швидко зникаючого полум’я, що супроводжується легким вибухом, допускається розходження між двома паралельними визначеннями плюс мінус 4˚С.
Температура самозаймання нафтопродукту визначають після визначення температури спалаху. Для цього продукт продовжують нагрівати з тою швидкістю (4˚С за 1 хв), через кожні 2˚С підносять до поверхні тигля полум’я пальника чи тліючу скибку. Відмічають температуру при, якій продукт займається і горить не менше 5 с, цю температуру приймають за температуру самозаймання продукту. Розходження між двома паралельними визначеннями повинно бути не більше 6˚С.
Визначення температури кипіння
Температура кипіння – це також індивідуальна характеристика рідини, як і температура плавлення. Найпростіший спосіб визначення температури кипіння це нагрівання аналізуючої речовини в приладі. Пари киплячої речовини конденсуються на шарику термометра, який протягом деякого часу показує постійну температуру.
В лабораторіях використовують інший спосіб визначення температури кипіння, аналізуючи рідину поміщають в тонку скляну трубочку запаяну з одного кінця і прикріплюють до термометра. В рідину опускають запаяний зверху капіляр, при наближенні до температури кипіння з капіляр починають виділятися окремі бульбашки повітря, які при досягненні температури кипіння переходять в рівномірний потік малих бульбашок.
Урок № 83-84 тема: Методи визначення умовної в’язкості (по швидкості витікання з віскозиметра типу Энглера), фракційного складу нафтопродуктів (перегонкою на стандартному приладі). Методи проведення випробування простих лакофарбових продуктів (визначення густини, в’язкості, кольору). Методи визначення лужного середовища. Правила користування універсальними індикаторним папірцем, кольоровість її відповідно до еталона. Ознайомлення з методами проведення хімічного аналізу вуглецевих і низьколегованих сталей на загальний вміст вуглецю в сплавах (метод спалювання наважки аналізую чого сплаву в атмосфері кисню в трубчастих печах). Метод газового аналізу: абсорбційний (метод поглинання) і метод спалення. Використання газоаналізатора типу Орса для аналізу газових сумішей. Шляхи поліпшення якості проведення аналізів. Охорона праці при виконанні аналізу рідких, газоподібних і твердих речовин.
В’язкість
– це властивість рідини здавати опір взаємному переміщенню руху її частинок. Від в’язкості нафтопродуктів залежать експлуатаційні властивості зношення деталей, які труться, від вод тепла від них і витрати масла. З підвищенням температури в’язкість зменшується і сильно зростає при пониженні температури. Ці зміни характеризуються індексом в’язкості, що являє собою температурний коефіцієнт в’язкості. Для цього визначення спів ставляють в’язкість масла при різних температурах (50˚С, 100˚С). Чим менше в’язкість залежить від температури тим вищий індекс. Розрізняють три види в’язкості:
- динамічну (абсолютну);
- кінематичну;
- відносну.
Динамічна
– визначається як сила в Динах, яка необхідна для взаємного переміщення із швидкістю 1 см/с двох шарів рідини, поверхнею 1 см, який знаходиться на віддалі на 1 см один від одного. Її вимірюють в Куазах чи санти Куазах. В системі СІ це Н*С/м.
Кінематична
– це відношення динамічної до густини при тій же температурі. Вимірюють в Стоксах чи санти Стоксах.
Відносна
– на практиці її визначають відношенням часу витікання досліджуваного продукту, до часу витікання води при тій же температурі. Її виражають градусами чи секундами. Число градусів Енглера
– відношення часу витікання іх віскозиметра Енглера 200 мл досліджуваного продукту при даній температурі до часу витікання 200 мл дистильованої води. Для перерахунку кінематичної в’язкості умовно користуються формулами і таблицями.
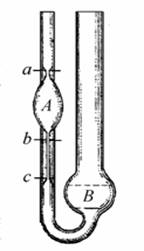
Визначення кінематичної в’язкості віскозиметром Оствальда
Принцип дії віскозиметра оснований на витіканні стовпчика досліджуваної рідини під дією сили ваги. Являє собою скляну у-подібну трубку в коліно 1 впаяний капіляр6, який з’єднаний з кулеподібним резервуаром 4. Контрольні мітки 3 і 5 служать для спостереження часу витікання певного об’єму нафтопродукту. Коліно 2 і резервуар 7 служать для наповнення віскозиметра аналізуючою рідиною.
Хід визначення
В добре вимитий, висушений віскозиметр вводять піпеткою певну кількість зневодненого і профільтрованого нафтопродукту, якщо аналізують дуже в’язкий нафтопродукт то віскозиметр заповнюють засмоктуванням його через коліно 1. Для цього віскозиметр перевертають і коліно 1 занурюють в посудину з досліджуваною рідиною за допомогою гумової груші засмоктують через коліно 2 досліджувану рідину, якщо вона дуже густа то її підігрівають. При наповненні слідкують щоб не було пухирців повітря. Далі його встановлюють у термостат де витримують протягом 30 хв при заданій температурі. Коли рідина дійде до мітки 3 включають секундомір і зупиняють його тоді, коли рідина пройде нижню мітку,віднімають час витікання рідини від верхньої до нижньої мітки. Вимірювання проводять не менше 5 разів. Беруть середнє значення. При роботі слідкують за тим, щоб на шариках і капілярах не утворювалась плівка, яка порушує режим витікання. = К*Т.
Визначення бромного числа полімеру.
Бромне число
– це кількість брому в грамах затраченого на приєднання до 100 г досліджуваного зразка.
Визначення кислотного числа
Кислотне число
– це кількість грамів лугу витраченого на титрування 100 г досліджуваного зразка.
Визначення естерного числа
його визначають для смол, масел, жирів. Для цього наважку досліджуваного зразка 0,5-1,2 г вносять в конічну колбу, розчиняють в 5 мл етилового спирту, добавляють 15 мл 1 н розчину NaOH, нагрівають на водяній бані 2 год, охолоджують і титрують 1 н розчином HCl в присутності фенолфталеїну. Молекулярну масу естеру вирахують:
M = 1000*g/C1*V1 – V2*C2
C1 – V1 – об’єм і концентрація кислоти;
C2 – V2 – об’єм і концентрація лугу.
Тема:
Фізико-хімічні (інструментальні) методи аналізу
Рівне - 2010
Урок № 85-86 тема:Особливості і галузі використання фізико-хімічних методів аналізу. Класифікація методів і їх характеристика. Оптичні методи. Фотометричні методи. Класифікація методів і галузі їх використання. Сутність хімічної теорії фотометричних методів.
Класифікація фізико-хімічних методів:
І електрохімічні
– основані на залежності між складом аналізуючої речовини і його електрохімічними властивостями.
- Кондуктометричний метод – оснований на здатності розчинів електролітів проводити електричний струм;
- Потенціометричний метод – використовується залежність між складом розчину і потенціалом, який виникає при зануренні в ньому електродів;
- Електроваговий метод – оснований на електролізі розчинів, осад, що виділився на електродах зважують і по ньому обчислюють кількість речовини, яка була в розчині;
- Полярографічний метод – оснований на електролізі розчинів і вимірюванні поляризації катода, яка відбувається в розчині під дією електро-окислення чи електровідновлення;
- Кулонометричний метод – оснований на вимірюванні кількості електрики використаної на електролізі аналізуючої речовини
В цих методах використовуються прилади кондуктометри, потенціометри, кулонметри, іономіри, солеміри, полярографи.
ІІ оптичні методи
– основані на оптичних властивостях аналізуючої речовини.
- Спектральні – основані на здатності атомів і молекул поглинати випромінювання з певною довжиною хвилі;
- Нефелометричний і турбодиметричний – основані на вимірюванні розсіяного світла твердими частинками;
- Фотоколориметричні і колориметричні – основані на вимірюванні інтенсивності світла, яке пройшло через аналізуючий розчин;
- Рефрактометричний – оснований на здатності різних речовин по різному заломлювати світло, яке проходить через них;
- Емісійний спектральний – оснований на здатності атомів сегмента в певних умовах випромінювати хвилі певної довжини.
Прилади колориметри, фотоколориметри, спектрофотометри, нефелометри, рефрактометри.
ІІІ хроматографічні методи аналізу
– основані на абсорбції і адсорбції. Розрізняють адсорбційно-розпридільчу, йонобмінну і осадкову хроматографію.
За агрегатним станом аналізуючої сумішшю розрізняють: газову і рідинну хроматографію.
За типом реакції розрізняють: окисно-відновну, осадкову, комплексоутворювальну.
За способом проведення: паперову, розпридільчу, колонкову.
Всі ці фізико-хімічні методи основані на проведенні аналітичних реакцій, кінець, яких визначають за допомогою приладів, які фіксують зміну фізичних властивостей речовин. Результат може записуватися на стрічці самописця, чи передаватись на світлове табло.
При аналізі фізичними методами взагалі не проводять хімічну реакцію, а тільки вимірюють параметр, що характеризує певну фізичну властивість аналізуючої речовини, заломлення чи розсіювання світла, поглинання чи випромінювання електромагнітних хвиль.
Радіометричні методи
– основані на використанні радіаційних ізотопів і вимірюванні радіоактивного випромінювання.
Мас-спектрометричні методи
– основані на визначенні мас окремих іонізованих атомів, молекул і радикалів після їх розділення в результаті комбінованої дії електричних і магнітних полів.
Електронного парамагнітного резонансу
– оснований на явищі резонансного поглинання деякими атомами, молекулами чи радикалами електромагнітних хвиль, прилад радіоспектрометр.
Ядерного магнітного резонансу
– в ньому використовується явище поглинання електромагнітних хвиль, обумовлене ядерним магнетизмом, прилад спектрометр ядерного магнітного резонансу.
Інструментальні методи аналізу використовують для регулювання технологічного процесу, і для аналізую чого контролю, а також науково-дослідницьких лабораторій.
Урок № 87-88 тема: Фотометричний метод. Основний закон фотометрії. Поняття про коефіцієнт пропускання і оптичної густини. Фотометр. Фотометричні візуальні (колориметричні) методи аналізу. Метод стандартних серій. Приклади коло метричних визначень.
Оптичні методи аналізу
В основі колориметричного методу аналізу лежать реакції утворення чи руйнування забарвлених сполук, тобто сполук, здатних поглинати світло. Інтенсивність забарвлення сполуки пропорційна концентрації розчину. В основі цього методу лежить хімічна реакція від якої залежить час витрачений на аналіз, чутливість і час методу. Його застосовують для визначення вмісту малих кількостей різних речовин (1*10 - 1*10 г в об’ємі 50-100 мл). Такі кількості не можна визначити ваговим та об’ємним методами.
При проходженні крізь забарвлений розчин монохроматичного пучка світла, частина його поглинається, а частина проходить крізь розчин, при цьому інтенсивність світла зменшується. Оптична густина розчину (А), яка дорівнює десятковому логарифму відношення початкової інтенсивності пучка світла (І0) до інтенсивності пучка світла (І), який пройшов крізь усю товщину (l) забарвленого розчину збільшується прямо пропорційно збільшення вмісту речовини.
Закон Ламберта-Бугера-Бера: залежність оптичної густини забарвленого розчину від концентрації речовини, товщини шару і молярного коефіцієнта поглинання
A = lg I0/I = E*l*C чи K*C*h
Поглинання монохроматичного світла пропорційне концентрації розчину і товщині шару.
В колориметричних методах в процесі вимірювання використовують стандартний розчин (це розчин порівняння, в якому вміст речовини відомий).
Порівняти два світлових потоки можна візуально
неозброєним оком, знімаючи концентрацію речовини, товщину шару чи інтенсивність світлового потоку, або за допомогою фотоелектричних приладів у яких світлова енергія перетворюється в електричний струм.
Метод стандартних серій
(шкали)
Інтенсивність забарвлення досліджуваного розчину порівнюють з інтенсивність забарвлення стандартних розчинів серії, для виготовлення якої беруть 10—15 однакових пробірок, у першу наливають 0,1 мл стандартного розчину, у другу — 0,2 мл і т. д.. збільшуючи його кількість за геометричною прогресією. Потім добавляють усі реактиви, потрібні для утворення забарвленої сполуки. Аналогічно обробляють досліджуваний розчин. Вміст речовини в досліджуваному розчині дорівнюватиме вмісту речовини в стандартному розчині з однаковим забарвленням.
Метод колориметричного титрування
Дві однакові пробірки діаметром 2—2,5 см і висотою 25—30 см вставляють у штатив, в обидві пробірки наливають реактиви. в першу — досліджуваний розчин, а в другу поступово добавляють стандартний (з відомою концентрацією речовини) розчин із бюретки. Стандартний розчин добавляють доти, поки інтенсивність забарвлення обох розчинів не зрівняється при однакових об'ємах. Розчини в обох пробірках весь час перемішують. Вміст речовини знаходять за об'ємом добавленого стандартного розчину. Це легко зробити, тому що концентрація стандартного розчину відома. Перевага методу колориметричного титрування перед методом шкали в тому, що цей метод можна застосувати тоді, коли забарвлена сполука недостатньо стійка в часі (тіоціанат феруму).
Колориметр
У колориметрах занурення зрівнюють інтенсивність забарвлення, змінюючи товщину шару розчину. Досліджуваний і стандартний розчини наливають у циліндричні скляні посудини, які за допомогою спеціальних механізмів можуть опускатись і підніматись. У ці циліндри вільно входять нерухомо закріплені скляні палички з оптичного скла. При опусканні чи підніманні циліндрів змінюється товщина шару забарвленого розчину, що фіксується на спеціальних шкалах, з'єднаних через покажчик рівня розчину з циліндрами. Однакова товщина шару в обох циліндрах при однаковій інтенсивності забарвлення обох половин поля зору свідчить про однаковість концентрацій обох розчинів. Однаковості поля зору можна досягти, змінюючи товщину шару одного з розчинів. Концентрацію досліджуваного розчину обчислюють за формулою:
Cx = Cст*lст/lx
lx – lст – товщина шару розчину.
Урок № 89-90 тема: Фотоелектрокалориметричний метод. Фотоефект, фотоелемент, фотоелектричний колориметр
У цьому методі за допомогою фотоелементів світлова енергія перетворюється в електричний струм. Метод оснований на вимірюванні оптичної густини розчину за допомогою приладів фотоколориметрів. Прилади побудовані по двохпроменевій схемі.
Оптична схема фотоколориметра
Світло від лампи накалювання за допомогою двох дзеркал ділиться на два паралельних світлових потоки однакової інтенсивності. Кожний світловий потік проходить через світлофільтр, кювету з аналізуючим розчином і розчин порівняння, попадає на фотоелемент, який перетворює світлову енергію в електричний струм. Електричний струм іде на вимірювальний прилад, який фіксує різницю електричних струмів. Інтенсивність світлових потоків урівнюють за допомогою оптичних клинків зв’язаних з відліковим барабаном і шкалою, в паралельному світловому потоці інтенсивність світла змінюють за допомогою щілевої діафрагми (котяче око). В момент рівності світлових потоків вимірювальний прилад встановлюється на 0 і по шкалі відлікового барабану знімають оптичну густину.
При роботі на фотоколориметрі використовують три кювети, у дві наливають розчин порівняння, а в третю аналізуючий розчин. Спочатку на шляху світлових потоків ставлять кювети з розчином порівняння, урівнюють інтенсивність світлових потоків тоді в правий потік ставлять кювету з аналізуючим розчином, переводять стрілку мікроампер метра до 0 і по правому відліковому барабану вимірюють оптичну густину.
Кювети виготовляють із кварцу чи з спеціальних сортів скла, вони мають певну товщину, яка враховується при розрахунках. Якщо розчин має темне забарвлення, то використовують тоненьку кювету, а для світло забарвлених розчинів товсту. При роботі використовують абсолютно чисті кювети, які беруть за ребра, так як найменші забруднення впливають на показники приладу. На шляху світлових потоків у фотоколориметрі ставлять світлофільтри, від 5 до 11, які пропускають певну частину спектра, вони мають різну довжину хвилі, їх підбирають для кожного визначення так: знімають оптичну густину при різних світлофільтрах і вибирають той світлофільтр при якому оптична густина буде максимальною. Вони являють собою скляні пластинки забарвлені в різні кольори, які пропускають промені певного спектра. При використанні жовтих розчинів беруть синій, червоних – зелений, для синіх – жовтий, для зелених – червоний.
Щоб визначити концентрацію речовин користуються калібрувальним графіком, який будують так: готують серію стандартних розчинів для кожного з них знімають оптичну густину, наносять на графік точки, які з’єднують і одержують градуювальник графік.
Вимірювання оптичної густини на фотоколориметрі ведуть двома способами:
І –
в правий пучок поміщають кювету з досліджуваним розчином, а в лівий з розчином порівняння. Лівий барабан встановлюють на 0, правим барабаном за допомогою оптичних клинків стрілку мікроампер метра встановлюють на 0. В правий пучок ставлять кювету з стандартним розчином, стрілка відхиляється від 0 за допомогою барабана ми ставимо її на 0 і по лівому знімаємо покази оптичної густини.
І –
в обидва пучки світла поміщають кювети з стандартним розчином. Правий барабан ставимо на 0, а потім стрілку мікроампер метра ставимо на 0. Правий барабан замість кювети з стандартним розчином ставлять кювету з досліджуваним розчином, стрілка відхиляється при цьому від 0 правим барабаном її знову ставлять на 0 і по шкалі правого барабана знімають оптичну густину. При виконанні аналізу притримуються якого не будь одного способу вимірювання.
Визначення речовин фото колориметричним аналізом. Визначення вмісту нікелю у водному розчині його солі
В фото колориметричному визначенні йонів нікелю оснований на його здатності утворювати забарвлені комплексні сполуки з реактивом диметилгліоксимом і окислювачем,для того щоб сполука була стійка, не руйнувалась необхідно дотримуватись порядку додавання реактивів: до солі нікелю додають окисник, диметилгліоксим спиртовий розчин і луг. Перед початком аналізу будують калібрувальний графік для цього готують серію стандартних розчинів з точно відомою концентрацією, знімають їх оптичні густини і будують графік залежності оптичної густини від концентрації. Так само до аналізуючої проби додають всі реактиви і знімають оптичну густину по відношенню до розчину порівняння, який містить ті самі реактиви крім нікелю. За оптичною густиною по калібрувальному графіку обчислюють вміст нікелю в аналізуючий пробі.
Визначення концентрації органічних барвників
Аналіз починаємо з побудови калібрувального графіка: для цього готуємо серію стандартних розчинів, які містять певну кількість барвника, залежно від властивостей барвника його розчиняють в дистильованій воді чи додають певну кількість соди чи кислоти. Деякі барвники взагалі не розчинні у воді, тому їх переводять в аерозолі чи суспензії, що містять дуже дрібні частинки і для яких зберігається пропорційна залежність оптичної густини від концентрації. Важливо правильно вибрати світлофільтр тому, що від цього залежить чутливість аналізу. Знімаємо оптичну густину стандартних розчинів і будуємо градуювальник графік.
Тому наважку аналізую чого барвника розчиняють в тих же умовах, що й стандартні розчини, знімають оптичну густину по калібрувальному графіку знаходять концентрацію аналізую чого барвника і обчислюють вміст барвника в наважці.
Визначення нітритів у стічних водах
Нітрити – це речовини, які шкідливо впливають на навколишнє середовище. Метод ґрунтується на властивості нітрогену нітритів утворювати в кислому середовищі і сульфаніловій кислоті діазосполуки, які реагують з L-нафтиламіном і утворює забарвленні яскраво-малинового кольору азосполуки:
HSO3C6H4NH2 + HNO2 + CH3COOH = HCO3C6H4N-NCH3COO + H2O
HSO3C6H4N-NCH3COO + C10H7NH2 = HCO3C6H4N-NC10H6NH2 + CH3COOH
Хід аналізу
В мірну колбу на 100 мл наливаємо від 1 до 50 мл аналізуючої води. Одночасно готуємо холосту пробу на дистильованій воді. В обидві проби добавляємо по 2 мл реактиву Гріса, доводимо до мітки дистильованою водою і перемішуємо. Колби з розчинами поміщають у водяну баню при температурі 50˚-60˚С на 10 хв. Після охолодження визначають оптичну густину із зеленим світлофільтром. Нульову точку встановлюють по холостій пробі, вміст нітратів визначають по калібрувальній прямій, розрахунок за формулою:
X = mнав*1000/VH2O мг/л
Урок № 91-92 тема: Порядок роботи, вибір світлофільтру і кювети, побудова калібрувальних кривих. Приклади кількісних визначень. Спектрофотометричний аналіз. Суть методу. Спектрофотометри, принцип їх дії. Оптичні схеми і будова приладів.
У спектрометрії використовують безбарвні розчини, вимірювання ведуть при хвилі певної довжині, для цього призма розкладає світло в спектр, вибираємо хвилю і проводимо аналіз. Щоб вибрати хвилю потрібної довжини то знімаємо оптичну густину одного і того ж розчину при різних довжинах хвилі. Вибираємо ту довжину хвилі при якій оптична густина буде максимальною.
Оптична схема спектрофотоколориметра
Поліхроматичне (біле) світло від лампи накалювання в області від 320 до 1100 нм чи водневої лампи, що дає світло в області від 190 до 320 нм, поступає на вхідну щілину дзеркала і призму, що розкладає світло на спектри. Світло відбивається від посрібленої задньої стінки призми. Відбивається від дзеркала поступає на вхідну щілину, яка вирізає вузьких монохроматичний участок спектра. Монохроматичне світло проходить через світлофільтр, кювету з розчином і попадає на фотоелемент, фотострум фотоелемента підсилюється підсилювачем і поступає на відліковий пристрій побудований за принципом компенсації фотострумів. Спектрофотометри можуть бути побудовані по одно- чи дво-променевій схемі.
Марки спектрофотометрів.
Спектрофотометр СФ-16
Спектрофотометр СФ-10, СФ-14
Реєструючий електрофотометр «Хитачи» 124
Спектрофотометр VSU-2-P
Спектрофотометр Спекол
Спектрофотометр Спекорд UV-VIS,71-IR,71-IR, Бекмана 34/35 UV/VIS
Правила роботи на спектрофотометрі
В кюветну камеру поміщають кювету з розчином порівняння і кювету з аналізуючим розчином. Обертаючий барабан довжин хвиль, встановлюють на певну довжину хвилі: спочатку в світловий потік поміщають кювету з розчином порівняння і за допомогою регулятора ширини щілини приводять до нуля стрілку мікроампер метра. Далі в світловий потік поміщають кювету з аналізуючим розчином і приводять вимірювальний прилад до нуля. Розрахунок концентрації розчину за даними оптичної густини одержаної на спектрофотометрі проводять за допомогою калібрувальних графіків чи за законом Ламберта-Бугера-Бера, якщо відома величино молярного поглинання. В цьому випадку користуємось формулою:
С=D*P/a*E*b
а – наважка речовини;
Р – розведення;
Е – питомий показник заломлення;
b – товщина шару.
При використанні молярного показника заломлення користуємось формулою:
С=D*М*P/a*E*b*10
М – молярна маса речовини;
Е – молярний показник заломлення;
Урок № 93-94 тема: Приклади кількісних визначень речовин і ідентифікація органічних сполук. Нефелометрія і турбодиметрія. Явища світлозсіювання і світло поглинання. Нефелометри. Приклади кількісних визначень. Поняття про спектрофотометричне і турбодиметричне титрування
Якщо світловий потік направити через дисперсну систему, частина світла розсіюється твердими частинками і світловий потік послаблюється, на цьому явищі основані нефелометричний і турбодиметричний методи аналізу.
При нефелометричнрму аналізі вимірюють інтенсивність потоку розсіяного світла Ір. Вимірювання ведуть під кутом 90˚ по відношенню падаючого світлового потоку.
Інтенсивність світлового потоку, розсіяного маленькими частинками, пропорційне концентрації твердих частинок у суспензії :
Ір=І0*К*С
Отже, чим більша концентрація завислих твердих частинок тим більша інтенсивність розсіяного світла. Для двох суспензій з частинками однакових розмірів і форм, інтенсивність розсіяного світла будуть відноситися між собою як концентрації розчинів:
Ірст/Ір ан=Сст/Cан=>Cан=Ір*Сст/Ір ст.
Вимірюючи інтенсивність розсіяного світла для аналізуючої суспензії з точною концентрацією, можна визначити концентрацію аналізую чого розчину.
При турбодиметричному аналізі вимірюють інтенсивність світлового потоку, який пройшов через кювету і ослабленого за рахунок розсіювання і поглинання світла суспензією. Залежність інтенсивності світлового потоку, що пройшов через суспензію від концентрації вираховують за формулою:
Lg=І0/І=К*С*l; D=R*C*l
Аналіз методом нефелометрії і турбодиметрії виконують за допомогою фотоелектроколориметрів-нефелометрів, чи нефелометрів з візуальним спостереженням через окуляр. Кількісний аналіз поводять за допомогою попередньо побудованого калібрувального графіка. Ці методи придатні для аналізу дуже розбавлених суспензій,які містять не більше 100 мг/л.
Порядок роботи
Визначення проводять по калібрувальному графіку, який будують за стандартними розчинами. Для проведення вимірювання встановлюємо вибраний світлофільтр, відлікові барабани ставлять на нуль по шкалі знімають оптичну густину. Далі поворотом дика підбирають такий розсіювач, при якому яскравість половин поля зору окуляра будуть найбільш близькі один до одного. Після наповнення кювети аналізуючим розчином поворотом правого барабану знаходять положення фотометричної рівноваги і проводять по ньому відлік оптичної густини, показник повинен бути не більше 0,15 – 0,25 так як при більшій густині зменшується точність вимірів, вимірювання повторюють 3-4 рази і за цими результати знаходять середнє значення. Необхідно зберігати чистоту стінок кювети, вода яка береться у кюветну камеру не повинна мати незважених частино, має бути дуже чистою.
Урок № 95-96 тема: Рефрактометрія. Закони відбиття і заломлення світла. Прилади для визначення показника заломлення. Принцип дії, оптичні схеми та будова приладів
Вона основа на вимірюванні показника заломлення аналізуючого розчину. Кожна індивідуальна речовина характеризується певним показником заломлення. Якщо промінь світла переходить із одного прозорого середовища в інше, то промені заломлюються.
Відношення синуса кута падіння до синуса кута заломлення називається показником заломлення:
n=sinL/sinB
Показник заломлення залежить від природи речовини і довжини хвилі падаючого світла, температури. Джерелом випромінювання служить натрієва лампа, для включення якої потрібний спеціальний пристрій. З підвищенням температури показник заломлення рідин зменшується, тому вимірювання ведуть при температурі 20˚С і в довідникових таблицях приводять показник заломлення виміряний при цій температурі.
Кут падіння L завжди менший від кута заломлення B, якщо L збільшувати, то наступає момент коли В дорівнює 90˚, тобто промінь не входить в друге середовище, а ковзає по його поверхні. Наступає повне внутрішнє відбивання світла
і кут падіння називається граничним кутом
:
n2*sinL=n1*sinB; n2*sinL=n1*sin90˚/1; n1=n2*sinL
Якщо відомий показник заломлення одного середовища досить виміряти граничний кут, щоб визначити показник заломлення другого середовища. На цьому принципі основані роботи на рефрактометометрі. Як середовище з відомим показником заломлення використовують призми із спеціальних сортів скла з високим показником заломлення. Відомо багато рефрактометрів, основаних на вимірюванні граничного кута.
Рефрактометр типу АББ, моделі УРЛ, ІРФ-22. У рефрактометрі цього типу світловий потік направляється на призму не безпосередньо, а через додаткову призму, яку називають освітлювальна
. Каплю аналізуючої рідини поміщають на поверхню однієї із прим і стискають призми. В зазорі між ними утворюється тонкий шар анлізуючої рідини 0,1-0,2 нм. Промінь світла від джерела світла входить в освітлювальну призму, заломлюється в ній, падає на шар аналізуюочої рідини і через неї на поверхню вимірювальної призми за якою визначаємо показник заломлення. Величину показника заломлення визначають після перевірки приладу за дистильованою водою (nводи=1,3330).
Визначення концентрації речовин в розчині ведуть за графіком, який будують визначаючи показники заломлення для серії стандартних розчинів. Крім графіків для розрахунку концентрації можна використовувати формулу:
Х=n-n0/F
n – показник заломлення розчину;
n0 – показник заломлення розчинника;
F – фактор, що показує збільшення показника заломлення при рості концентрації на 1%;
X – концентрація розчину %.
Аналіз меду
Сутність методу: він ґрунтується на залежності показника заломлення меду від вмісту в ньому води.
Хід аналізу
Для проведення визначення використовують рідкий мед. В разі, якщо мед закристалізований, поміщають 1 см його в пробірку, щільно закривають гумовим корком і нагрівають на водяній бані при температурі 60˚С до повного розчинення кристалів. Тоді охолоджують до кімнатної температури, інтенсивно розмішують скляною паличкою.
Першу краплю меду наносять на призму рефрактометра і вимірюють показник заломлення. Отриманий показник заломлення меду перераховують на масову частку води у меді. Допустимі розходження між результатами контрольних визначень не повинні перевищувати 0,1 %. Згідно стандарту масова частка води повинна становити 19-21%.
Урок № 97-98 тема: Фотометрія полум’яна. Характеристика методу та галузь застосування. Принципові схеми полум’яного фотометру. Приклади якісних і кількісних визначень
Вона основана на вимірюванні світлової енергії у полум’ї. При фотометрії аналізуючий розчин стиснутим повітрям чи киснем у вигляді аерозолю вводять в полум’я газового пальника, при наявності в розчині йонів легко збуджуваних елементів, полум’я забарвлюється в наслідок характерних випромінювань, які фіксуються фотоелементом. Фотострум, який виникає вимірюється чутливим мікроамперметром. Величина фотоструму залежить від концентрації речовини в полум’ї, від складу полум’я, температури, спання дисоціації сполук на атоми і ступеня іонізації атома у полум’ї.
Полум’яна фотометрія широко застосовується у кольоровій металургії, при аналізі різноманітних рід. В наш час цим методом визначають більше 50 елементів.
Будова приладу
Досліджуваний розчин за допомогою стиснутого повітря подають в розпилювач, звідки він у вигляді аерозолю, попадає в полум’я пальника. Випромінювання полум’я збирається ввігнутим дзеркалом і направляється лінзою на світлофільтр, а далі до фотоелементу. Фотострум, що виникає підсилюється підсилювачем і вимірюється чутливим мікроампер метром.
Вимірювання натрію в розчині
Аналізуючий розчин наливають у мірну колбу на 100 мл і доводимо дистильованою водою до мітки, перемішують. Готують серію стандартних розчинів. Для цього в мірні колби на 100 мл додають 0,3; 0,4; 0,5; 0,6; 0,7 мл натрію. Ці розчини готують на бідистильованій воді. Їх наливають в стакани, з яких повітрям подають в полум’я пальника і вимірюють показники. На основі цих даних будують калібрувальний графік. Після цього в полум’я вводять аналізуючий розчин, знімають показник по калібрувальному графіку знаходять концентрацію. Вимірювання починають з розчину з найбільшою концентрацією. Після кожного вимірювання розпилювач і газовий пальник ретельно промивають водою. Кожне вимірювання проводять не менше 3 разів.
Урок № 99-100 тема: Електрохімічні методи. Класифікація електрохімічних методів і галузі їх використання. Методи електролізу. Ступінь електрогравіметричного аналізу. Установка для електрогравіметричного аналізу. Кулонометрія. Основи методу – закони Фарадея. Кулонометрія при постійному потенціалі. Кулонометрія при постійній силі струму. Умови введення електролізу. Схеми установки для кулонометричного аналізу. Схеми установки для кулонометричного аналізу. Приклади кількісних визначень
Електроваговий метод аналізу
Метод оснований на електролізі розчинів. Для електролізу використовують електроди (платинові), бо до них ставлять такі вимоги:
· Електроди не повинні вступати в реакцію з аналізуючою речовиною;
· Не повинні окислюватися киснем, повітрям.
Таким вимогам відповідають платинові електроди, осад що утворився, повинен щільно прилягати до електродів.
Визначення міді в розчині сульфату міді
Електроваговий метод визначення міді полягає на відділенні міді з кислого розчину на платиновому катоді за допомогою електролізу:
Сu+2е=Сu
Катодом служить платинова сітка анодом-платинова спіраль. Для цього їх поміщають на димний гас в гарячий розчин 1:1 нітратної кислоти і промивають дистильованою водою. Катод крім цього обробляють спиртом, ефіром висушують і витримують у ваговій кімнаті деякий час, після чого зважують на аналітичних вагах. Складають прилад: джерелом струму служить свинцевий акумулятор, який дає напругу приблизно 2В. Беруть чистий стакан на 150 мл в який поміщають розчин сульфату купруму, який містить 0,1-0,15 купруму. Приливають до нього 7-8 мл 2н розчину нітратної кислоти і 3 мл розбавленого розчину сульфатної кислоти (1:4). Потім опускають в стакан платиновий сітчатий електрод(катод) і закріплюють його так, щоб не торкався дна стакана і стінок. Другий – електродоплатинову спіраль(анод) – закріплюють так, щоб він був у центрі сітчатого електрода. Після цього потрібно розбавити аналізуючий розчин водою, щоб рівень рідини в стакані був приблизно на 1 см нижче верхнього краю сітки. Стакан нагрівають скляними пластинками. Для того, щоб розчин не розбризкувався. Потім сітчатий катод приєднують до негативного, а спіраль-анод до позитивного полюсу джерела струму. Розчин підігрівають слабким полум’ям пальника не вище 60̊ С, бо Сu почне розчинятися. Електроліз потрібно продовжити до тих пір, поки розчин знебарвиться , після того потрібно провірити повноту осадження міді якісною реакцією з К4(FeCN6).Переконавшись в повноті осадження міді приступають до промивання електродів, не виключати струм. В стаканах на 150 мл з дистильованою водою швидко переносять електроди. Склянку з водою піднімають на стільки, щоб електроди були повністю зануренні у воді. Через 1-2 хв промивання повторюють. Слідкуючи за тим, щоб електроди залишались на повітрі всього декілька секунд. Міняють воду 3-4 рази до припинення виділення на аноді бульбашок. Після цього включають струм. Катод промивають спиртом, ефіром і швидко висушують над стінкою, чи у муфельній печі з закритими дверима. Після першого зважування катод потрібно ще раз пропустити і зважити до постійної маси. По різниці мас катода до і після електролізу визначають кількість купруму.
Кулонометричний аналіз
Оснований на вимірюванні кількості електрики витраченої на електроліз. При прямому кулонометричному аналізі аналізуючи речовина реагує безпосередньо на електродах. При цьому використовують такі типи хімічних реакцій:
- нейтралізації;
- осадження;
- окислення відновлення;
- комплексоутворення.
Для виявлення кінцевої точки титрування використовують індикатори, або електрохімічні методи. Завдання полягає в точному визначенні кінця титрування, визначення наявності електрики, яка пішла на титрування і визначення вмісту аналізуючої речовини.
Для кулонометричного титрування складають установку в яку входять:
- джерело струму;
- реостат;
- мікроамперметир;
- перемикач;
- опір потенціометра;
- комірка;
- генераторний електрод;
- індикаторний електрод;
- електрод порівняння;
- допоміжний електрод;
- амперметр.
В основі цього методу лежать закон Фарадея із якого визначають масу проредагованої речовини:
m=1/f*A/n*Q
m – кількість проредагованої речовини;
n – кількість відомих чи приєднаних електронів;
f – число Фарадея 96500 кл – електрики чи 26,8 А год;
Q – кількість електрики;
A – атомна маса розчину.
Кулонометричний аналіз використовують в тих випадках, коли в результаті електролізу утворюються як розчинні так і нерозчинні сполуки. Електроліз ведуть при малому струмі, кількість речовини, що визначається повинна бути малою, щоб аналіз не забрав багато часу і був точним.
Кулонометричне титрування
Воно використовується тоді, коли аналізуючи речовина не бере участь безпосередньо в аналітичній реакції. В аналізуючий розчин добавляють допоміжний реагент, який окислюється чи відновлюється на електродах, а потім реагує з аналізуючою речовиною. Відбувається титрування аналцзуючого розчину тим реактивом, який утворився в результаті електролізу. За кількістю витраченого реагента судять по кількості електрики, яка витрачена на електроліз, так проводиться титрування.
Урок № 101-102 тема: Кондуктометрія. Сутність методу. Електропровідність розчинів, її залежність від концентрації. Апаратура для вимірювання електропровідності розчинів. Кондуктометричне титрування
Вона основана на вимірюванні електропровідності розчинів. Розрізняють еквівалентну
і питому електропровідність
.
Питома електропровідність
– це електропровідність розчину, який міститься між плоскими електродами, що знаходяться на віддалі на 1 см один від одного.
Еквівалентна електропровідність
– це питома електропровідність 0,1 н розчину в електроліті.
Електропровідність розчину крім концентрації залежить ще від продуктивності йону електроліту. Чим швидше рухається йон, тим більший електричний струм проходить через розчин. Кондуктометричний аналіз проводять за допомогою кондуктометрів
– приладів, які вимірюють опір розчину. По величині опору легко визначити електропровідність розчину, яка вимірюється:
L=1/R Ом
Кондуктометри (солеміри) побудовані по мостовій схемі да в комірку з розчином опускають електроди, які вмонтовані в плече моста. Міст складається із реохорда
– натягнута дротина, по якій рухається бігунець постійного опору R, і вимірювального «нуль приладу» поміщеного в діагональ моста. До точок А і В підключають електричний струм і пересувають движок реохорда, знаходять точку в якій вимірювальний прилад покаже відсутність напруги (нуль). В цей момент опір комірки Rx так відноситься до постійного струму Rc, як опір плеча реохорда (дротина Lx) до опору плеча Lc:
Rc/Lc=Rx/Lx Rx=Rc*Lx/Lc
Знайшовши Rx по формулі L=1/R знаходимо L, а по калібрувальному графіку визначаємо концентрацію розчину.
Кондуктометричне титрування
Його застосовують головним образом при реакціях нейтралізації, осадження і комплексоутворення. За допомогою кондуктометричного титрування можна визначити речовини, які не можна виконати звичайними способами об’ємного аналізу: аналіз суміші сірчаної і соляної кислот, суміші хлоридів і бромідів.
Точний об’єм аналізую чого розчину поміщають в електролітичну комірку і вимірюють його електропровідність. Результати титрування вносять в таблицю і будують графік кондуктометричного титрування: по осі абсцис відкладають об’єм прибавленого робочого розчину, а по осі ординат – електропровідність. Побудована таким чином крива кондуктометричного титрування показує зміни електропровідності розчину в ході титрування. В точці еквівалентності характер кривої змінюється переломом чи вигином кривої. Вміст аналізуючої речовини визначають за формулою:
Nx=V*N/Vx
Vx – об’єм аналізую чого розчину, взятого для аналізу, мл;
Nx – концентрація аналізую чого розчину, г*екв/л;
V – об’єм робочого розчину витраченого на титрування до ТЕ, мл;
N – концентрація робочого розчину.
Урок № 103-104 тема: Потенцірметрія. Сутність методу і галузі його використання. Залежність потенціалу електроду від концентраціх іонів. Індикаторний електрод і електрод порівняння. Іонометрія, рН-метрія. Іоноселективні електроди
Цей метод оснований на вимірюванні електричного потенціалу, який виникає на електродах опущених в розчин з аналізуючою речовиною. Електричний потенціал виникає на електродах реакції окислення відновлення, його величина Е визначається рівнянням Нернста:
Е=Ео + 0,059/n lg [окисник]/[відновник]
Ео – стандартний потенціал;
n – число електронів.
Величина стандартного потенціалу окислення відновлення визначається по відношенню до Н електрода, вимірюється зі зміною потенціалу водневих іонів в розчині. В потенціометричному аналізі використовують скляні електроди, потенціал, яких залежить від потенціалу водневих іонів. Скляний електрод складається із скляної трубки із шариком на кінці. В трубці 0,1 н розчин хлориду калію і срібна дротина.
Для проведення потенціометричних визначень в аналізу чий розчин потрібно опустити два електроди робочий і електрод порівняння, потенціал якого постійний. В якості робочого електроду в потенціометрії використовують скляний електрод. Порівняльним електродом служить каломейний чи хлор срібний електрод. Каломейний електрод являє собою скляну посудину на дно якої налита ртуть, поверх ртуті знаходиться паста із каломею Hg2Cl2 і налитий насичений розчин хлориду калію. В ртуть опущена дротина для контакту з приладом. Хлоро срібний являє собою скляну трубку в якій запаяна срібна дротина, покрита шаром хлориду срібла і занурена в розчин хлориду калію. Контакт електрода з розчином відбувається через листок із азбестової нитки змоченої хлорижом калію.
На практиці для вираження кислотності чи лужності розчинів замість концентрації [H] використовуємо її водневий показник рН
рН=lg[H]
Буферний розчин
– це розчин із строго встановленим значенням рН.
Індикаторне визначення рН
визначають наближено з допомогою індикаторного приладу. Значення рН досліджуваного розчину знаходять так: кольори спів ставляють зі шкалою порівняння.
Визначення концентрації водневих іонів
Для визначення використовують скляні електроди. Електродом порівняння використовують хлор срібний чи каломейні електроди. Щоб зменшити похибку зв’язану з потенціалом новий скляний електрод витримують у воді, чи в 0,1 н розчині хлоридної кислоти протягом 1-2 діб. Перед вимірюванням попередньо коректують шкалу рН приладу за буферними розчинами. Для цього в скляну наливають буферний розчин занурюють в нього скляний і каломейний електроди і вимірюють рН. Вибирають той буферний розчин значення рН, якого близьке до рН аналізую чого розчину. Встановлюють відповідну температуру, реохорд встановлюють на рН буферного розчину при цьому стрілка гальванометра відхиляється в сторону, її встановлюють в нульове положення обертанням рукоядкику компенсатора. Відкоректувавши таким чином прилад по буферному розчині, приступають до вимірювання рН. Розчин поміщають в склянку занурюють в нього електроди, попередньо промивши їх дистильованою водою і включають прилад, по шкалі якого знімають покази рН.
Рідше для вимірювання рН розчинів використовують сурм’яний, хіргідронний електроди.
Урок № 105-106 тема: Потенцірметричне титрування. Потенціометри. Приклади потенціометричних визначень
Потенціометричне титрування полягає в тому, що точку еквівалентності при титруванні визначають за різкою зміною різниці потенціалів пари електродів, які поміщені в аналітичному розчині. Найчастіше в методі використовують такі реакції:
- осадження;
- окислення відновлення;
- нейтралізації.
Реакції, які використовуються при потенціометричному титруванні повинні бути практично не оборотними, протікати з великою швидкістю і тільки в певному напрямі, а в точці еквівалентності повинна проходити помітна зміна потенціалу індикаторного електрода (скачок потенціалу). Потенціометричне титрування проводять так: в аналізуючий розчин занурюють електродну пару і після додавання певної порції титр анта вимірюють електродний потенціал. Результати титрування вносять в таблицю і будують криві титрування, на осі абсцис відкладають об’єм добавленого розчину, а на осі ординат електродний потенціал (мВ). В точці еквівалентності плавний характер кривої змінюється. По ньому визначають витрати робочого розчину на реакції. Розрахунок ведуть за звичайною формулою об’ємного аналізу:
V1*C1= V2*C2 C(x) = C1*V1/Vx
Урок № 107-108 тема
: Полярографія. Сутність методу. Явище концентраційної поляризації. Граничний чи дифузійний струм. Вольт-амперна крива. Принципова схема полярографічної установки. Полярографи. Зняття полярограм. Амперометричне титрування. Теорія методу і його використання. Криві амперметричного титрування
Цей метод оснований на вимірюванні сили струму, яка змінюється від напруги в процесі електролізі, при умові що один із електродів (катод) має дуже малу поверхню, а інший (анод) велику поверхню. Сила струму (граничний дифузійний струм) пропорційна концентрації аналізую чого розчину в речовині.
Принципова схема полярографа
1. Пристрій для подачі потенціалу.
2. Полярографічна колонка
3. Регістратор.
Суть методу полярографії:
метод оснований на вимірюванні величини струму, який виникає при відновленні чи окисленні речовини на електродах. Якщо в розчин з речовиною опустити два електроди і прикласти потенціал, то на електродах при певному потенціалі почнуться електрохімічні процеси окислення на аноді і електро-відновлювання на катоді, це явище називається поляризацією.
В поляризації використовують два види електродів:
І – ртуть налита на дно посудини;
ІІ – ртутний капельний електродний.
Ртутний капельний електрод, який являє собою товстостінну капілярну трубку з’єднану з резервуаром для ртуті. Із капіляра з постійною швидкістю витікають крапельки ртуті. Поверхня електроду оновлюється, що гарантує чистоту поверхні, відсутність забруднення постійної площі. Електрод із великою поверхнею служить шар ртуті на дні комірки чи каломейний електрод Hg2Cl2. Ділянка полярографа на якій відбувається підвищення струму називається полярографічною хвилею
. Допустимий струм, який визначає швидкість дифузії називається дифузійним струмом
. Величина потенціалу середньої точки хвилі називається потенціалом півхвилі
. Потенціал півхвилі є характеристикою речовини. Кожна речовина має певний потенціал хвилі –
якісний полярографічний аналіз. Висота хвилі зв’язується з концентрацією речовини.
Якщо речовини в розчині мало, то полярографічна хвиля має певну висоту, при збільшенні концентрації речовини висота хвилі росте. Залежність дифузійного струму І від концентрації С описується рівнянням Іньковича
І = К*С К = 607u D * M *r
Кількісний полярографічний аналіз
І – метод калібрувальних аналіз
Полягає в тому, що змінює полярограму ряду точних розчинів і наносимо в систему координат. Будуємо калібрувальний графік, яким користуємось при визначенні концентрації аналізую чого розчину.
ІІ – метод стандартних розчинів
Знімаємо полярограму аналізую чого розчину, а потім будуємо полярограму стандартного розчину близької концентрації до аналізуючої. По відношенню висоти хвилі стандартного і аналізую чого розчину обчислюють концентрації аналізую чого розчину:
Cст/hст=Cан/hан Сан = Сст*hан/hст
ІІІ – метод добавок
Знімають спочатку полярограму аналізуючого розчину, потім додають до нього точну кількість визначаючої речовини і знову знімають полярограму. По збільшенню висоти хвилі в результаті додавання вираховують вміст визначаючої речовини в розчині:
Cх/hх=Cдоб/hдоб Сх = Сдоб*hх/hдоб
Амперометричне титрування
Основане на тому, що кінцеву точку титрування знаходять по зміні сили граничного дифузійного струму, який проходить через розчин при сталій напрузі між індикаторним електродом і електродом порівняння. За результати титрування будують графік залежності струму і об’єму робочого розчину на кривій знаходять точку перетину двох віток кривої, яка відповідає кінцевій точці титрування, тобто вимірюють об’єм розчину, який витратився на титрування і за основною формулою об’ємного аналізу проводять розрахунки.
Урок № 109-110 тема: Методи розподілу і концентрування. Класифікація і характеристика методів розподілу. Екстракція як метод розподілу, її сутність. Величини, які характеризують процес екстракції. Техніка екстрагування. Роль екстрагування в підвищеній чутливості і селективності визначень
Хроматографія як метод розподілу і аналізу речовин. Сутність методу і галузі використання. Основні поняття. Класифікація методів хроматографії в залежності від способу переміщення сорбатів вздовж шару сорбенту. Характеристика елюційного, фронтального витискаючого методу і електрохроматографії. Класифікація методів хроматографії в залежності від механізму сорбції. Характеристика адсорбційної іонообмінної, осадкової, розпридільчої і гель-хроматографії. Класифікація методів хроматографії в залежності від агрегатного стану рухомої і нерухомої фаз. Характеристика газової хроматографії та її варіантів: газо-абсорбційної і газорідинної хроматографії. Сутність рідинної хроматографії і її варіантів: рідинно-рідинної, рідинно-адсорбційної хроматографії. Класифікація методів хроматографії в залежності від техніки хроматографічного розподілу. Характеристика колон очної паперової і тонкошарової хроматографії. Класифікація методів хроматографії в залежності від мети проведення хроматографічного аналізу.
Методи основані на явищі абсорбції і десорбції. Хроматографічні методи основані на неоднаковій здатності компонентів по різному поглинатись і виділятись, при проходженні суміші через абсорбент. Компоненти проходять через колонку, ті що поглинаються гірше переміщаються потоком далі по колонці, а ті що сорбуються міцніше відстають. Процес сорбції і десорбції повторюються багаторазово. Розрізняють такі види хроматографії:
- Адсорбційна
- Осадкова
- Розпридільча
- Окисно-відновна
- Комплексоутворююча
- Іонообмінна
- Паперова
- Газова хроматографія.
Адсорбційна хроматографія
– основана на вибірковій адсорбції окремих компонентів суміші твердим порошкодібним адсорбентом (силікагель, активоване вугілля, молекулярні сита).
Осадкова хроматографія
– основана на утворенні осадів при проходженні аналізуючої речовини через колонку з адсорбентом.
Розпрдільча хроматографія
– основана на використанні процесів розприділення речовин між двома розчинниками, що не змішуються один з одним. Закон розприділення:
Kp=C1/C2
С1 – концентрація речовини в розчині,
С2 – концентрація речовини в розчиннику.
Виконується колонковим, паперовим і тонкошаровим методами, папір N 1–2 швидкий, папір Nc 3-4 повільніший.
Окисно-відновна хроматографія
– основана на неоднаковій швидкості окисно-відновних реакцій, що протікають між речовинами, які містяться в колонці і іонами хроматографуючого розчину.
Адсорбційна комплексоутворююча
– основана на використанні різних величин констант нестійкості комплексних сполук, що утворились.
Йонообмінна хроматографія
– основана на обміні йонів між розчином і адсорбентом. Адсорбенти
– це іонообмінні смоли, які являють собою неорганічні чи органічні сполуки, що містять активні групи здатні обмінюватись на іони, катіони, аніони. Катіообмінні смоли володіють рухомими катіонами і обмінюють їх на катіони з аналізую чого розчину. Вони містять активні групи COOH, SO3H, OH. При взаємодії розчину, який містить катіони металів з кат іонообмінною смолою проходить така реакція:
RCOOH + K = RCOOK + H
Катіонообмінні смоли марки КУ-1 та КБ-4.
Іонообмінні смоли – це анааніти містять активні групи, які здатні обмінюватись на аніон. NO3 , NH3OH та інші. Реакція йоного обміну протікає за рівнянням:
RNH3OH + An = RNH3An + OH
Марки аніонітів АВ-17, АН-2.
Катніоніти настоюють при перемішуванні з 3% розчином хлоридної кислоти протягом 12 год, а потім промивають дистильованою водою. Аніоніти залишають на 12 год в №% розчині хлоридної кислоти, промивають дистильованою водою, обробляють 2-4 год 2% розчином NaOH і промивають водою. Сорбентом заповнюють колонку для хроматографування, на дно поміщають тампон, або скляну пористу пластинку, заповнюють адсорбентом з дистильованою водою і зверху тампон.
Визначення кухонної солі
(NaCl)
Пропускають через колонку 5-10 мл (точно аналізуючого розчину) рідину, що витікає, збираємо в колбу разом із промивними водами. Одеожаний розчин титруємо лугом, проводимо розрахунки.
RCOOH + HCl = RCOONa + HCl
HCl + NaOH = NaCl + H2O
Урок № 1
11
-11
2
тема:
Апаратурне оформлення процесу газової хроматографії. Системи реєстрування. Хроматограми. Техніка хроматографії. Методи обчислення хроматограф. Застосування газової хроматографії для автоматизації виробничих процесів. Вимоги з охорони праці при виконанні аналізу
Газова хроматографія
Принципова схема газового хроматографа: газохроматографія основана на різній властивості компоненту поглинатись і виділятись при проходженні через абсорбент. Вся система безперервно продувається газом носієм (H2N2CO2) із балона. Проба аналізую чого газу вводиться в газовий потік за допомогою пристрою для вводу газу. Газ носій просуває суміш через колонку і детектор, в колонці газова суміш розділяється на складові компоненти. Пропускаючи в детектор, який фіксує (виявляє) їх , падає сигнал, який записує на стрічці автоматичного реєстратора.
Принцип дії детектора по теплопровідності катетометра
Має дві камери – камери порівняння туди поступає чистий газ-носій, і вимірювальна камера туди поступає газ-носій з аналізуючою пробою. Поки через дві камери іде чистий газ носій, вимірювальна схема і реєструючий прилад пером самописця на паперовій стрічці записує нульову лінію, коли із колонки потоком газу-носія виноситься в детектор перший компонент аналізуючої проби, рівновага виміряної суміші порушується, сигнал поступає на записуючий пристрій і фіксується пером самописця, як відхилення від нульової лінії – поява піку.
Способи розшифровки хроматограф
Основні складові хроматографа: кришка, азбестова ватка, металічний корпус, вигоди терморегулятора, ізолюючі втулки, газовий пальник.
Обчислювання результатів аналізу проводять по хроматографі по порядку і часу виходу піків встановлюють якісний склад суміші. Хімічний склад суміші визначають, по площі чи висоті піку. Площу обчислюють, як добуток на висоту та ширину. Обчислюють площу кожного піку, потім знаходять їх суму, а від суми знаходять процент до кожного піку, що відповідає вмісту визначаючих компонентів. Для багатьох газів існує коефіцієнти на яких
Sст/Cст = Sан/Cан Caн = Сст*Sан/Sст
hcт/Cст = hан/Cан Cст = Cан*hан/hст
Тема:
Технічний аналіз
Рівне - 2010
Урок № 1
1
3-114 тема: Технічний аналіз неорганічних речовин. Його значення у виробничо- технічному оцінюванні сировини і визначення хімічних, фізичних і експлуатаційних властивостей кінцевих продуктів виробництва. Методи технічного аналізу
Технічний аналіз
– це сукупність хімічних, фізичних і фізико-хімічних методів дослідження сировини напівпродуктів і готової продукції, їх відповідність, встановленим нормам і стандартам.
Методи технічного аналізу: хімічні, фізичні, фізико-хімічні.
Види технічного аналізу залежно від об’єкту контролю і мети аналізу розрізняють маркерувальні і швидкісні аналізи. Маркерувальні аналізи проводять для дослідження хімічного складу і властивостей матеріалів, які поступають на підприємництво (завод). Це дуже важливе завдання так як тільки про використанні доброякісної вихідної сировини можна забезпечити виробництво стандартної продукції при мінімальних затратах сировини, палива і робочої сили. Маркерувальний аналіз також призначений для об’єктивної оцінки результатів роботи підприємництва. По результатам аналізу визначають якість напівпродуктів і готової продукції, її відповідність встановленим нормам. Маркування аналізу повинні відрізнятися високою точністю, так як на основі даних аналізу виконують технологічні, а також фінансові розрахунки. До маркувальних аналізів також відносять арбітражні аналізи.
Арбітражні аналізи за своїм характером являється контрольними аналізами. Вони виконуються третьою незацікавленою стороною. При виконанні претензій споживача до постачальника в результаті розходження результатів аналізу, які визначають якість продукції. Арбітражні аналізи виконують висококваліфіковані робітники, при чому, як правило тими ж методами, що й маркерувальні.
Швидкісні-експресні аналізи проводять для внутрішнього заводського контролю і спостереження за протіканням процесу підприємництва на найбільш відповідальних стадіях може забезпечити прийняті режими роботи і відповідно нормальний вихід продукції, яка відповідає стандарту. Крім необхідної точності ці аналізи повинні визначатися швидким виконанням.
Урок № 117-118 тема : Аналіз води. Показники контролю якості води: сухий залишок, твердість, завислі частинки, окислюваність, лужність кислотність вміст хлоридів, сірчистих сполук, азотовміістких сполуки. Методика визначення
Показники контролю якості води:
Органолептичні показники якості води:
- Колір – залежить від наявності в ній органічних і неорганічних домішок;
- Запах – за походженням може бути пов'язаний з наявністю в ній живих і мертвих організмів, вплив берегів і дна, з потраплянням у воду інших речовин та об’єктів;
- Смак – вода має бути приємною на смак, освіжаючою, що зумовлено розчинними в ній мінеральними солями і газами. Неприємний смак чи присмак зазвичай залежить від великого вмісту в воді деяких солей і органічних речовин;
- Присмак – буває гірко-солоний, кисло-солоний, гірко-солодкий;
- Прозорість – визначається кількістю завислих в ній речовин. Чим більше мінеральних і органічних речовин у воді, тим вона каламутніша. Дуже каламутна вода малопридатна до споживання без попередньої обробки. Вона може спричинити шлунково-кишкові захворювання.
Фізичні показники води:
- Температура – вимірюють комбінованими чи максимальними термометрами. Вона залежить від температури навколишнього середовища, а також від низки умов і від походження і глибини водних джерел. У відкритих і мілких водоймах температура води протягом року змінюється, тоді як у глибоких підземних джерелах вона стала;
- Густина – залежить від вмісту в ній органічних і мінеральних сполук. Її вимірюють за допомогою ареометра.
Хімічні показники води: лужна реакція води, сухий залишок, твердість, наявність розчинного кисню, аміаку, нітритів і нітратів, хлоридів, сульфітів феруму.
Реакція води.
Вода, забруднена органічними речовинами тваринного походження і продуктами гниття, часто має лужну реакцію, а вода, забруднена стічними водами промислових підприємств — кислу. Кислу реакцію мають води болотного походження, кислотність яких зумовлена наявністю нешкідливих органічних гумінових кислот. Добра вода повинна мати нейтральну або слабколужну реакцію (рН 6,5-8,0). Кисла або лужна реакція вище згаданої норми свідчить про забруднення водного джерела.
Сухий залишок
— густий залишок, одержаний при випаровуванні одного літра профільтрованої води, свідчить про загальну кількість розчинених у воді речовин мінерального та органічного походження. Б добрій воді залишок світло-сірого або білого кольору, тоді як сухий залишок води, забрудненої органічними речовинами або сполуками феруму і мангану, має жовто-буре або темно-буре забарвлення.
Густий залишок може бути показником мінералізації води. Для визначення загальної кількості мінеральних речовин у сухому залишку води його прожарюють. Сухий залишок води не повинен перевищувати 1000 мг/л. Слід зазначити, що прямої залежності між кількістю сухого залишку і забрудненням води немає.
Твердість води
зумовлюють наявні в ній солі кальцію і магнію (Са і Мg) здебільшого вуглекислі та сірчанокислі. Тверда вода небажана для господарських і технічних потреб. У ній погано прати, бо збільшується витрата пральних засобів, погано розварюються плоди. Тверда вода утворює на стінках котлів твердий накип, зменшуючи їхню теплопровідність до 15% і більше. Перехід від м'якої води до твердої, особливо якщо вона містить багато сульфатів магнію (МgSO4
), при напуванні тварин часто спричиняє розлад шлунково-кишкового тракту (проноси). М'яка вода також небажана для напування тварин, оскільки вона не забезпечує їх необхідними солями, тому тварини п'ють її неохоче. Твердість води визначають у повних одиницях — градусах твердості. Останнім часом її визначають в умовних одиницях — градусах твердості. Нині твердість визначають у міліграм-еквівалентах на літр води. Один міліграм-еквівалент твердості відповідає наявності 20,04 мг Са або 12,16 мг Мg у літрі води (де 20,04 і 12,16 — еквівалентні маси Са і Мg, рівні половині їх атомних мас). Твердість доброї води має відповідати 7 мг-екв./л, іноді допускається до 14-18 мг-екв./л.
Вода з твердістю до 10° м'яка, від 10 до 20' — помірно тверда, понад 20° — тверда. Твердість питної води повинна становити не більше 30-40', для тварин можна використовувати і твердішу воду.
Окиснюваніеть води.
У воді з різних джерел можуть міститись різні органічні речовини рослинного і тваринного походження, а також мікроорганізми. Наявність у воді великої кількості органічних речовин свідчить про забрудненість води в санітарному відношенні. Кількість органічних речовин у воді визначають непрямим методом за потрібним для окиснення киснем. Звідси, чим більше у воді органічних речовин, тим більше кисню витрачається на окиснення, тим більша окиснюваніеть води. Але слід зазначити, що при аналізі не повністю окиснюють-ся органічні речовини, водночас можуть частково окиснитися деякі мінеральні сполуки (нітрати, сульфати І закис заліза). У зв'язку з цим окиснюваніеть води дає тільки уявлення про наявність у ній легко окиснюваних речовин, не вказуючи на їхню природу.
Окиснюваніеть води коливається у великих межах. Так, у глибоких підземних водах (артезіанських свердловинах, джерелах і глибоких шахтних колодязях) окиснюваніеть становить 1-2 мг/л. У воді неглибоких шахтних колодязів і відкритих проточних водойм окиснюваніеть може сягати 4 мг/л, а у воді непроточних водойм (озера, ставки) — 6-8 мг/л. У болотних водах окиснюваніеть зазвичай становить 8-20 мг/л. Окиснюваніеть доброї питної води не повинна перевищувати 2-5 мг/л кисню.
Розчинений кисень.
До складу води входять розчинений кисень, який потрапляє з повітря. За кількістю розчиненого у воді кисню можна визначити наявність у ній органічних речовин. Чим чистіша вода, тим більше в ній кисню. У воді відкритих водойм кисень постійно використовується на окиснення органічних речовин. Через це у дуже забрудненій воді розчиненого кисню може не бути зовсім. У воді відкритих водойм при середній температурі (10-20'С) кисню міститься 5-20 мг/л. Глибокі підземні води кисню не мають, але дуже швидко збагачуються ним на повітрі. Під час оцінки води часто з'ясовують біохімічне використання кисню (БВК). З цією метою визначають зменшену кількість розчиненого кисню після п'ятидобового зберігання проби води при температурі 20'С. Чим більше досліджувана вода містить органічних речовин, тим меншою буде концентрація розчиненого кисню.
Аміак, нітрити (солі нітритної кислоти) і нітрати (солі нітратної кислоти).
Альбуміноїдний аміак є продуктом розпаду білкових речовин тваринного походження. Сольовий аміак може бути і мінерального походження як продукт відновлення азотнокислих солей під впливом денітрофікуючих бактерій. Його знаходять у воді, де є гумінові кислоти і в незабрудненій воді. Нітритна кислота є продуктом початкової стадії окислення аміаку.
Нітрити і нітрати
. Деяка кількість нітратної кислоти може уворюватись у дощовій воді під дією електричних розрядів під час грози. В цьому разі наявність у воді нітритної кислоти не є показником її забруднення. Вміст у воді альбуміноідного аміаку, а також солей аміаку та нітритної кислоти свідчить про забруднення її органічними речовинами тваринного походження (гній, сеча тощо) і робить таку воду дуже шкідливою в санітарному відношенні. Велика кількість аміаку та нітратної кислоти в питній воді може бути причиною отруєння тварин, особливо молодняку. Описано багато випадків водно-нітритної метгемаглобінемії у дітей, особливо там, де концентрація нітритів у питній воді перевищує ЗО мг/л. У доброякісній питній воді аміаку і нітритної кислоти не повинно бути зовсім або тільки у вигляді залишків. Наявність солей не тільки нітратної кислоти (за відсутності аміаку і солей нітритної кислоти) свідчить про те, що процес окиснення (мінералізація) завершився і така вода не шкідлива. Якщо водночас з солями нітратної кислоти у воді є аміак і солі нітритної кислоти, то де джерело забруднення.
Хлориди.
У воді хлор трапляється у формі хлоридів (КаСІ, К.С1, МgСІ2, СаСІ2). Велика кількість хлоридів у воді буває в разі забруднення її сечею, гноївкою, стічними водами або у воді, що тече по солончакових ґрунтах, багатих на хлористі сполуки. Так, є місцевість з солончаковою водою, яка містить 300-500 мг/л хлоридів. Така вода не шкідлива і придатна для господарсько-питного використання. Якщо велика кількість хлоридів не пов'язана з солончаковим Грунтом, супроводжується великою окиснюваністю, присутністю аміаку і солей нітритної кислоти, така вода недоброякісна і непридатна для напування тварин. Допустиму кількість хлоридів у питній воді встановлюють залежно від походження хлориду — тваринного або мінерального.
Сульфати,
або солі сульфатної кислоти, з'являються у воді під час окиснення білкових речовин, які мають сульфур. Але сульфати можуть бути у великій кількості в незабрудненій воді, наприклад, якщо вона містить гіпс. Наявність сульфатів у воді непостійна, їхня кількість значно коливається, що залежить від біологічних умов окремих районів держави. Наприклад, у деяких районах вміст сульфатів у воді буває 2000-3000 мг/л. Санітарне значення сульфатів таке само, як і хлоридів. Вода з великим вмістом сульфатів натрію (Ш28О4) і магнію (М&804) гірка на смак і має послаблювальну дію, спричиняє розлад діяльності шлунково-кишкового тракту у тварин.
Ферум (залізо).
Наявність великої кількості феруму в воді не є показником забруднення, але це різко змінює її органолептичні показники.
Вода з великим умістом феруму непридатна для використання в молочному виробництві, так як вона надає молоку, вершкам і маслу поганого присмаку, а у виробах утворюються Іржаві плями. При пранні такою водою білизни на ній також залишаються іржаві плями. У водопровідних трубах ця вода сприяє розмноженню залізобактерій, що може призвести до закупорювання просвіту труб.
Дослідженнями встановлено важливу роль для організму тварин мікроелементів, які є в воді. Це стосується фтору, вміст у воді якого (1,5-2 мг/л) призводить до захворювання людини і тварин, яке називають флюорозом. З іншого боку, дуже низький вміст фтору б питній воді (менше 0,4 мг/л) спричиняє карієс зубів, особливо у дітей. Доведено, що нестача йоду в питній воді є основною причиною виникнення ензоотичного зобу у тварин. Дослідами встановлено токсичну дію води, в якій у великій кількості є плюмбум, арсен, меркурій, барій та інші речовини внаслідок потрапляння їх у відкриті водойми зі стічними водами промислових підприємств. У воді відкритих водойм і шахтних колодязів інколи знаходять залишкову кількість гербіцидів групи сечовини, гептахлору та пестицидів. Тож необхідно досліджувати воду на наявність і цих речовин.
Отже, показники хімічного аналізу можуть свідчити про безпечну або шкідливу питну воду в санітарно-токсикологічному відношенні, а також про її фізіологічну цінність. Шкідливу дію на людей і тварин має вода, забруднена радіоактивними речовинами. З метою дотримання норм і правил радіоактивної гігієни відповідні лабораторії повинні здійснювати дозиметричні дослідження води.
Вимоги до питної води
Основні вимоги щодо питної води:
| Показник
|
Норма
|
| Калорійність, не більше
|
20
|
| Присмак і запах при температурі 20°С, балів
|
2
|
| Прозорість, см, не менше
|
30
|
| Загальна твердість, мг-еке./л
|
7
|
| Окиснюваність, мг/л
|
5
|
| Сухий залишок, мг/л
|
1000
|
| Вміст, мг/л, хлоридів
|
350
|
| сульфатів
|
500
|
| феруму
|
0,3
|
| купруму
|
1
|
| цинку
|
5
|
| мангану
|
0,1
|
| аміачного нітрогену
|
Сліди
|
| нітрогену нітритів
|
Сліди
|
| нітрогену нітратів
|
10
|
| Колі-титр
|
300
|
| Колі-індекс
|
3
|
| Кількість бактерій у 1 мл (мікробне число)
|
100
|
До джерел водопостачання належать різні природні води.
Атмосферні води
— це дощова і снігова вода. Атмосферні води утворюються в результаті конденсації парів. Ця вода близька до дистильованої. Вона має дуже мало солей і розчинених газів, м'яка, без смаку. В атмосферній воді є органічні речовини, мінеральний пил і мікроорганізми, які потрапляють із повітря під час проходження її через атмосферу. Дощова вода, зібрана над лісовими масивами і полями, має менше пилу, мікроорганізмів і різних хімічних домішок. Снігова вода часто буває поганої якості, оскільки внаслідок тривалого лежання сніг дуже забруднюється. Атмосферні води використовують для напування тварин тільки у безводних районах.
Наземні води.
До наземних або відкритих водойм належать: річки, озера, ставки, лимани, водосховища, моря, болота.
Річкова вода
бере початок від атмосферних, болотних, озерних і джерельних вод, а також від розтавання снігу і льоду.
Озера
— водойми зі стоячою водою.
Водосховища
— це штучні водойми великих розмірів, утворені внаслідок зарегулювання греблями долин річок, виходів із озер, гірських потоків і щілин. Водою вони поповнюються насамперед у період весняних повеней.
Вода боліт
повністю непридатна для напування тварин і для іншого вжитку у зв'язку з великою забрудненістю її органічними речовинами, а також мікроорганізмами і яйцями гельмінтів. Така вода загниває, «цвіте» і, як засвідчує ветеринарна практика, спричинює різні захворювання тварин.
Підземні води
— це води, які залягають на різних глибинах земної кори. Вони утворюються внаслідок фільтрації атмосферних і поверхневих вод у глиб землі.
Урок № 119-120 тема : Аналіз газів. Значення аналізу газів в різних галузях промисловості. Методи аналізу газів. Характеристика абсорбційного методу аналізу. Газоаналізатори, принцип їх роботи. Схема газоаналізаторів. Приготування поглиначів. Хроматографічний метод аналізу газових сумішей
Методи аналізу газів різноманітні і основані на хімічних чи фізичних властивостях газів:
- Термохімічний метод газового аналізу – оснований на вимірюванні теплового ефекту хімічної реакції;
- Віскозиметричний метод оснований на вимірюванні в’язкості газів (прилад віскозиметр);
- Денсиметричний метод – оснований на вимірюванні густини газів (прилад денсиметр);
- Волюмометричний метод (газооб’ємний) – оснований на скороченні об’єму газової проби при поглинанні окремих газових компонентів рідкими чи твердими поглиначами. Його використовують при відносно великих концентраціях компонентів газової суміші;
- Конденсаційний метод – оснований на конденсації аналізуючої газової суміші, при низьких температурах, його використовують для аналізу багатокомпонентних газових сумішей;
- Сорбційні методи – до них належать хроматографічні, які основані на абсорбції компонентів газової суміші;
- Інтерферометричні – оснований на різниці коефіцієнтів заломлення газів.
Промислові гази поділяються на 4 групи:
1. горючі газові суміші – природний газ, бензол, аміак, генераторні гази, колошниковий газ (газ доменних печей);
2. гази які використовують як хімічну речовину – етилен, кисень, азот, газ колчеданих пече, азотоводнева суміш;
3. відпрацьовані гази – димові гази різних промислових печей, хімічних та інших виробництв (димові гази – це продукти згоряння палива, вони містять азот, кисень, вуглекислий газ, водяні пари, які викидаються в атмосферу);
4. повітря промислових приміщень (атмосферне повітря).
Газоаналізатор Орса (ГХП-3М)
Робота цього приладу основана на явищі абсорбції, тобто поглинання із газової суміші компонентів рідкими чи твердими поглиначами, в результаті взаємодії між ними з утворенням нових речовин. Для кожної складової частини газової суміші застосовується специфічний поглинач. В якості твердого поглинача вуглекислого газу натронне вапно, рідким поглиначем цього газу застосовують 20-30% гідроксид калію. Для поглинання СО (чадного газу) застосовують мідно-аміачний (250гр NH4Cl розчиняють в 750мл води і добавляють 200гр CuO2). Рідким поглиначем для кисню є лужний розчин пірогалолу (C6H3(OH) 3 10гр його розчиняють у 30мл дистильованої воді і розбавляють до 200мл гідроксидом калію).
Перед початком роботи в урівнюючи склянку 6 наливають запираючу рідину (запираюча рідина – це слабкий розчин хлоридної кислоти в якому є індикатор метилоранж, тобто розчин рожево-червоного кольору). Запираючу рідину насичують аналізуючим газом, для цього аналізуючий газ набирають в бюретку і видалять з приладу, поглинаючі розчини об’ємом до 200мл наливають через відростки 8 у відповідні поглинаючі посудини. В сорочку бюретки 3 наливають розчин лугу, у другу наливають пірогалол, а в третю нітроаміачний розчин. Перед початком роботи необхідно перевірити герметичність частин приладу і щільність кранів. Для цього вхідний кран відкривають і піднімаючи зрівнювальну склянку 6 заповнюють бюретку 14 запираючою рідиною до мітки у верхній частині бюретки. Закривають вхідний кран і спускають склянку 6. Якщо рівні рідин в поглинаючих склянках і бюретці спочатку трішки спустяться, а потім залишаються постійними – прилади герметичні. Якщо не герметичні - то вхідний кран чистять і змазують вазеліном, або замінюють.
Перед аналізом рівні рідин в поглинаючих склянках потрібно доводити до мітки, які знаходяться у капілярах. Для цього поглинаючу посудину в якому має бути піднятий рівень розчину поворотом крана з’єднують з бюреткою при закритих інших кранах. Опусканням склянки 6 в бюретці утворюється вакуум в результаті чого рівень розчину в посудині 12 починає підніматися, і коли він дійде до мітки кран закривають. Після підготовки поглинаючих посудин відкривають вхідні крани і піднімають урівнювальну склянку 6 на таку висоту, щоб рідина у бюретці 14 досягла верхньої мітки (100).
Для відбору проби газу прилад через у подібну трубку 1 з фільтруючим матеріалом з’єднують із джерелом газу, трьохходовий кран 3 ставлять у положення «закрито» і продувають фільтр аналізуючим газом, потім кран відкривають і опускають в зрівнювальну склянку затягуючи у бюретку 100мл затягую чого газу. Закривають кран із суміщенням рівнів замираючої рідини в склянці і бюретці зрівнюють тиск газу з атмосферним, після цього записують початковий об’єм газу V1. Спочатку поглинають вуглекислий газ, для подачі газу в поглинаючу посудину 3 з розчином лугу, піднімають склянку 6 на стільки, щоб рівень рідини в ній перевищував рідини в бюретці, відкривають кран 11 поглинаючої посудини 3 і підйомом зрівнювальної склянки 6 переводять пробу газу в поглинаючу посудину. Для повноти поглинання вуглекислого газу, газ декілька разів повертають в бюретку і переводять знову в поглинаючу посудину, після декількох перекачувань газ залишають в бюретці, вимірюють об’єм V2 , різниця об’ємів (V1- V2) відповідає об’єму вуглекислого газу. Далі в такій же послідовності поглинають кисень в другій поглинаючій посудині 2 з пірогалолом V3 і СО чадний газ в першій посудині мідно аміачним розчином V4.
Xco2 = (V1- V2)*100 / V1
Xo2 = (V2- V3)*100 / V1
Xco = (V3- V4)*100 / V1
Урок № 1
21-
12
2
тема : Аналіз твердого палива. Склад палива. Вміст баласту в різних видах палива. Приготування аналітичних проб. Визначення вмісту вологи, зовнішньої і аналітичної. Визначення вмісту золи. Визначення вмісту загальної сірки. Визначення виходу летючих речовин. Визначення теплотворної здатності палива за даними елементного аналізу та колориметричним методом. Сутність колориметричного методу. Обладнання колориметричної установки, виконання аналізу
До твердого палива відносять вугілля, торф, горючі сланці і дрова. Тверду частину палива без вологи називають органічною масою палива
. Вона включає в себе карбон, водень, кисень, азот, частину сірки. Мінеральна частина палива
– це суміш неорганічних речовин (силікатів лужних і лужноземельних металів, залізо-амонію, сульфатів і сульфідів цих металів). Робоче паливо
– це паливо в якому стані воно поступає. Воно складається із горючої і негорючої маси, негорюча називається баластом
– це мінеральні речовини, волога, кисень, азот все це називається золою
.
Визначення вологи
. Вона є зовнішня і аналітична. Зовнішня волога у вигляді тонкої плівки, яку можна виділити підсушуванням речовини до температури 50˚С в сушильній шафі до постійної маси. Аналітична волога складається із адсорбційної вологи (називається гігроскопічною) і визначається висушуванням повітряної сухої наважки до постійної маси в сушильній шафі при температурах 100-110˚С. Зважену наважку з точністю 0,1 г (500 г вугілля), 200-300 г торфу, чи дров у вигляді стружок. Поміщуємо в сушильну шафу нагріту до 50˚С. Пробу бурого вугілля і торфу сушать на протязі 5 год, а кам’яне вугілля 3 год. За час сушіння пробу перемішують 3-4 рази. Пробу торфу після сушки витримують 6 год на повітрі, потім знову сушать і зважують. Сушку всіх видів палива проводять до тих пір поки різниця двох послідовних зважувань буде не більше 1 г. Розрахунок:
W(палива) = g1-V2 / g *100
V(палива) = g1-V2 / g *100
Визначення золи
. Золою
називаються негорючі залишки палива після спалювання. При спалюванні палива відбуваються такі перетворення: силікати втрачають свою кристалогідратну воду, солі оксидів заліза перетворюються у Fe2O3, карбонати розкладаються з виділенням вуглекислого газу, пірит окислюється до SO2, гіпс втрачає кристалогідратну воду.
Метод полягає в оголенні проби палива в муфельній печі при 850˚С. Пробу вугілля з масою 1г (точно)поміщають у фарфорову лодочку і прожарюють в муфельній печі при 850˚С протягом 2 год, охолоджують на повітрі 5 хв, а потім в ексикаторі і зважують, проводять контрольне прожарювання протягом 30 хв, до тих пір поки різниця двох послідовних зважувань буде не більше як 0,0004 г. Для розрахунків беруть останній результат і вміст золи визначають за формулою:
X = Q1 / Q *100
Q1 – маса зольного залишку,
Q – наважка палива.
Визначення теплотвірної здатності палива
. Теплотвірна здатність
– це тепло, яке виділяється при згорянні 1 кг палива. Це тепло затрачається на нагрівання води колориметричної бомби, мішалки та інших частин колориметра. Чим більша ця здатність тим краща якість палива.
Визначення сірки
. Метод полягає в спалюванні наважки палива і суміші Елека (MgO + Na2SO3) розчиненні утворених сульфатів і осадження їх хлоридом барію у вигляду сульфату барію, який фільтрують, зважують, прожарюють і проводять розрахунки.
Урок № 123-124 тема: Аналіз металів та сплавів. Характеристика основних сплавів. Загальні методи аналізу металів і сплавів. Легуючі домішки. Методики аналізів на вміст основних легуючих елементів у сплавах на основі титану, нікелю, вольфраму, кобальту, молібдену і ніобію
До металів відносять речовини, які мають такі характерні властивості: висока теплопровідність і електропровідність, металевий блиск, непрозорість, пластичність, ковкість.
Розрізняють чорні метали (залізо і його сплави), кольорові (алюміній, срібло, магній). Технічно чисті метали містять домішки. Кольорові метали в більшості випадків використовують у вигляді сплавів. Найбільш поширеними є латунь, бронза, дюралюміній, меркурій.
Сталь - це сплав заліза з вуглецем в якому вуглецю міститься 1,7%. Залежно від вмісту вуглецю розрізняють такі марки сталей. Сплав заліза з вуглецем з вмістом більше 1,7 називається чавуном. Чавун - твердий, але крихкий і не піддається куванню, чи прокату. Для покращення властивостей чавуну його легують додаючи добавки кольорових металів, які покращують якість чавуну.
Визначення вмісту вуглецю у сплаві.
Присутність вуглецю погано впливає на якість сплавів. Вуглець в сплавах може знаходитися в чотирьох формах:
- у вигляді вільного вуглецю (графітового чи аморфного вуглецю)
- у в’язкому стані у вигляді різних карбідів
- у вигляді твердого розчину
- у вигляді газоподібної форми (оксид вуглецю, водню)
Загальний вміст вуглецю визначають методом спалювання на важки металу чи сплаву в атмосфері киснем, в трубчастих печах при температурі 1250-1400˚С. При цьому весь вуглець окислюється до вуглекислого газу, який визначають газооб’ємним, об’ємним, або потенціометричним методом.
Суть газооб’ємного методу полягає в тому, що утворений вуглекислий газ направляють у газо-вимірювані бюретки, де вимірюється об’єм газу. Далі газ пропускають через розчин лугу, де проходить його поглинання:
2KOH + CO2 = K2CO3 + H2O
Після цього знову вимірюють об’єм газу, по різниці визначають об’єм вуглекислого газу, по якому обчислюють вміст вуглецю в сплаві.
Об’ємний метод використовують для визначення загальної кількості вуглецю. Утворений вуглекислий газ після його спалювання поглинають титруванням розчину лугу:
Ba(OH)2 + CO2 = BaCO3↓ + H2O
Надлишок розчину гідрооксид барію відтитровують янтарною, оцтовою, соляною кислотою по фенолфталеїну:
Ba(OH)2 + 2CO3COOH = Ba(CH3COO)2 +H2O
Потенціометричний метод дуже подібний до об’ємного і дає точні результати.
Ваговий метод полягає в тому, що рідкі поглиначі вуглекислого газу замінені на тверді (гідрооксид калію поміщають в дугоподібну трубку, яку зважують до поглинання і після поглинання, різниця мас дає вміст вуглекислого газу за яким визначають вміст вуглецю в сплавах):
KOH + CO2 = KHCO3
2KOH + CO2 = K2CO3 + H2O
Методи визначення сірки.
Сірка шкідливо впливає на якість сталі, визиває її красну ломкість, підвищує здатність до корозії, понижує кислотостійкість. Вміст сірки в чавуні 0,02-0,08%, в сталях ще менше. Для визначення сірки існує кілька методів:
1. метод відгонки – пробу розчиняють в спеціальній склянці з хлоридною кислотою, при цьому виділяється сірководень, який кількісно поглинають аміачним розчином, хлоридом кадмію або сульфідом цинку. Одержані сульфіди обробляють хлоридною кислотою при цьому виділяється сірководень який титрують розчином йоду в присутності крохмалю:
FeS + 2HCl = FeCl2 + H2S
H2S + I2 = S + 2HI
2. фотолометричний метод є найбільш точний, полягає в розчиненні наважки у фосфатній кислоті в присутності метилового хрому для відновлення SO4, S яку фотометрують на фотоколориметрі
Визначення фосфору.
Він негативно впливає на якість сплавів надаючи їм крихкість. Визначення полягає в окисленні фосфору до фосфатної кислоти сильним окисником (нітратна кислота, царська водка), яку можна визначити ваговим або коло метричними методами.
Ваговий метод оснований на осадженні фосфору у вигляді нерозчинного фосфорно- молібденового комплексу жовтого кольору. Аналіз закінчують осушуванням осаду при 110˚С і зважуванням.
Об’ємний метод полягає в розчиненні осаду у надлишку лугу, який відтитровують кислотою. Цей метод використовують для прискореного і маркерувального аналізу.
Колориметричний метод оснований на відновленні основної жовтої фосфорнр-молібденової кислоти в комплексну сполуку синього кольору – молібденову синь. Інтенсивність забарвлення фотокориметрують на фотоколориметрах, визначають оптичну густину, а по калібрувальному графіку знаходять концентрацію розчину.
Визначення легуючих елементів.
Нікель, хром, кобальт, манган додають до сталі, щоб покращити її якість. Концентрацію 3,4,8% нікелю в сплавах визначають об’ємним, ваговим, колориметричним і електро-ваговим методами.
Кобальт – придає сталям високу твердість і кислотостійкість. Ваговий метод полягає в осадженні кобальту органічним осаджувачем, утворений осад прокалюють при температурі 780-850˚С, зважують у вигляді CO3O4, проводять розрахунки. Його можна визначити об’ємним, фото колориметричним, полярографічним і потенціометричним методами.
Хром – визначають персульфатним, колориметричним, потенціометричним та іншими методами аналізу.
Визначення міді – є багато методів визначення міді, найголовнішими є електрохімічні, йодометричний, колориметричний, комплексонометричний, спектральний і полярографічний, найбільш поширений електро-ваговий метод аналізу.
Урок № 125-126 тема: Аналіз змащувальних матеріалів. Основні показники які характеризують склад і властивості мастил. Визначення низькотемпературних властивостей: температури застигання, помутніння, початку кристалізації. Визначення в’язко-температурних характеристик. Поняття динамічної, кінематичної і умовної в’язкості. Типи віскозиметрів. Визначення температур спалаху і займання, прилади. Аналіз нафти і нафтопродуктів. Нафта, її склад. Основні продукти нафтопереробки: паливо, змащувальні масла, сажа, бітуми та інші. Основні показники, які характеризують склад і властивості нафти і нафтопродуктів
Нафтопродукти розділяють на три основні класи:
І – паливо рідке і газоподібне;
ІІ – мастила;
ІІІ – нефтяні продукти промислового і побутового призначення.
Моторне паливо розділяють на три групи:
- карбюраторне (бензин, керосин);
- дизельне (для двигунів внутрішнього згоряння, керосин, солярка);
- котельне паливо для спалювання (мазут).
Кожен вид палива поділяється на марки властивості, яких повинні відповідати вимогам стандартів, в якому вказані числові значення основних фізико-хімічних властивостей: в’язкість, фракційний склад, кислотність, механічні домішки, вміст вологи і сірки, коксованість, температура самозаймання і застигання. За цими даними встановлюють сорт палива і його якість. Якість палива встановлюють за антидетонаційними властивостями, що оцінюють октановим числом.
Мастила
. Вони виконують такі функції: зменшують тертя між поверхнями, які рухаються, знижують їхній знос, безперервно очищають поверхню від різних механічних домішок, відводять тепло від деталей, що нагріваються, захищають від корозії, ущільнюють в циліндрах.
Щоб мастило могло виконувати такі функції воно повинно мати високу маслянистість, певну в’язкість, бути стабільним при зберіганні, бути термічно стійким не реагувати з киснем повітря, мати низьку температуру застигання і високу температуру спалаху, містити найменшу кількість органічних кислот, сірчистих сполук, механічних домішок і води при спалюванні утворювати мало золи. Залежно від умов роботи масла поділяються на:
1. індустріальні;
2. турбінні (компресорне, циліндрове, для підшипників) температура 150-200˚С;
3. масла для двигунів внутрішнього згоряння (авіаційні, автотракторні) температура 300˚С і більше;
4. трансформаторні масла;
5. трансмісійні (в коробках передач);
6. масла для холодильних машин;
7. спеціальні захисні масла.
До нафтопродуктів відносять густі масла ( більше 60 марок), універсальні (У) – вони є низько плавкі та високо плавкі.
Парафін
– це суміш твердих вуглеводнів від С19Н40 до С35Н72, використовуються в текстильній і паперовій промисловості, для ізоляції, в медицині, парфумерії, в кондитерських виробах. Він випускається різних сортів: високоочищений марки А і Б, медичний, очищений, не очищений.
Асфальти
– це нефтяні бітуми чорного кольору, тверді, добуваються із перегонки мазуту. Використовують для будівництва доріг, як покрівельний матеріал, електрохімічний матеріал. Найважливіший показник – температура розм’якшення.
Аналіз мастил.
В мастилах визначають густину (ареометром і пікнометром), в’язкість (віскозиметром), температура спалаху, температура застигання, температура самозаймання, вміст води (по методу Діна і Старка та методом відгонки), механічних домішок (фільтруванням), золи, мінеральних кислот чи лугів (титруванням).
Урок № 127-128 тема: Аналіз у виробництві неорганічних продуктів. Постадійний контроль виробництва. Точки контролю. Аналіз сульфатної кислоти і олеума. Визначення вмісту сульфатної кислоти. Визначення вмісту вільного сірчаного ангідриду
На кожній стадії виробництва ведеться контроль сировини, напівпродуктів, готового продукту. Для цього є точки контролю де відбирається проба для аналізу.
Аналітичний контроль виробництва H2SO4
1 стадія виробництва кислоти
- спалювання сірки чи випалювання колчедану:
- контактне окислення SO2 до SO3 на каталізаторі:
- абсорбція SO3 кислотою з утворенням олеуму:
Визначення сірки в сировині
Визначення вологи
визначають по зменшенню маси після висушування:
W = m–m1*100% / m
Визначення золи
20 г сірки точно зважують поміщають в попередньо прокалений до постійної маси тигль, сірку повільно спалюють, залишок прокалюють в муфельній печі при 800˚С до постійної маси і зважують. Розраховують: W = m1 / m
Визначення діоксину сірки йодометричним методом
При пропусканні газу, який містить діоксин сірки через розчин йоду проходить окислення діоксину сірки до H2SO4:
SO2 + I2 + 2H2O = H2SO4 + 2HI
По кількості взятого йоду і по кількості газу пропущеного через розчин визначають вміст діоксину сірки в газі, розрахунок вмісту діоксину сірки в газі визначають у об’ємних долях чи у відсотках по рівнянні:
SO2 = 10*K*1.095*100 / V0+10*K*1.095
Vg – об’єм витікшої води, приведеної до мілілітрів
V0 – об’єм 0,1 н розчину йоду взятого для аналізу
K – поправочний коефіцієнт, для приведення концентрації йоду до точно 0,1 н
1.095 – об’єм SO2, який відповідає 1 мл 0,1 н розчину йоду
Визначення ступеня контактування.
Це відношення SO2 до SO3. Для цього визначають вміст SO2 до контактного апарату і після нього йодометричним методом. Проводять розрахунки.
Аналіз готового продукту.
Визначення вмісту моногідрату – метод оснований на титруванні контактної сульфатної кислоти розчином гідрооксиду натрію в присутності метилового оранжевого:
H2SO4 + 2NaOH = Na2SO4 + 2H2O
Мірну колбу на 250 мл вливають 150 мл дистильованої води 5-6 г кислоти зваженої в бюксі з точністю до 0,0022, переносять в колбу, охолоджують до 20˚С і доводять до мітки. Перемішують відбирають піпеткою 20 мл цього розчину в конічну колбу і титрують 0,1 н розчином гідрооксиду натрію в присутності метилового червоного до переходу червоного забарвлення в жовте. Вміст сульфатної кислоти обчислюють за формулою:
W = V*K*0.025*250*100 / 25*m
V – об’єм гідрооксиду натрію витрачений на титрування
K – поправочний коефіцієнт
m – маса кислоти
0.025 – маса кислоти, яка відповідає 1 мл 0,5 н розчину NaOH
Аналіз олеуму. Визначення вільного
SO
3
в олеумі
Олеум чи димлячи H2SO4 являє собою розчин газу SO3 в 100% H2SO4. Склад олеуму характеризується вмістом вільного SO3 (більш 100%, яка містить в олеумі). У відповідності із стандартом він містить не менше 19% вільного SO3. Загальний вміст SO3 визначають титруванням NaOH, а далі по спеціальній таблиці знаходять вміст вільного SO3 в олеумі.
Хід визначення
В скляну ампулу на 2-3 мл з витягнутим капілярним кінцем зважують з точністю до 0,0002 г. Кульку ампули злегка нагрівають на слабкому полум’ї пальника. В ампулі утворюється розрідження, але цей гарячий капіляр опускають в аналізуючу кислоту і кислота взаємодіє з олеумом після цього кількість капіляра витирають фільтром зважують, визначають вміст відібраної проби олеуму.
Скляну банку на 400-500 мл наливають 150 мл дистильованої води поміщають кілька брусиків чи шматочків скляної палички, опускають ампулу з олеумом і щільно зактивають банку, перемішуємо вміст банки енергійно струшуючи, що продовжуємо до повного поглинання водою утвореного туману. Банку відкривають, роздавлюють скляною паличкою шматочки капіляра, зливають в банку розчин проби і додають 2-3 краплі метилового червоного і титрують NaOH. Вміст SO3 визначають за формулою:
W (SO3) = V*K*0.002*100 / m
V – об’єм NaOH витрачений на титрування
K – поправочний коефіцієнт до 0,5 н
m – маса кислоти
0.002 – маса відповідає 1 мл 0,5 н NaOH
За обчисленим вмістом SO3 по спеціальним таблицям знаходимо вміст вільного SO3 в олеумі. Вміст вільного SO3 в олеумі в масових долях, в % вираховують по формулі:
W (SO4) = (100*SO3)*98 / 18
98 – молярна маса H2SO4
18 – молярна маса води
Урок № 129-130 тема: Контроль у виробництві добрив. Визначення вмісту азоту в азотних добривах формальдегідним методом, методом Кєльдаля і нітрометричним методом. Визначення вмісту фосфору в фосфорних добривах цитратним та фото колориметричним методами. Визначення вмісту калію в калійних добривах, перхлорат ний метод
NH4OH – аміачна вода
NH4NO3 – аміачна селітра
(NH4)SO4 – сульфат амонію
KNO3 – індійська селітра
NaNO3 – чилійська селітра
Ca(NO3)2 – норвежська селітра
Є азотні, фосфорні і калійні мінеральні добрива. Для кожного добрива, що випускається є державний стандарт в якому зазначено правила прийому і відбору проби, методи аналізу, марки і технічні вимоги, упаковки, маркерування, транспортування та зберігання, гарантія виробки марки А і Б.
Контроль у виробництві азотних добрив
До азотних добрив відносять сульфат амонію, натрій, калій, аміачну селітру. Всі вони розчинні у воді і містять азот у зв’язаному вигляді. Виробництво азотних добрив повинно збільшуватись так, як в світовому масштабі, що річно вносять 25 млн тон зв’язаного азоту.
Сульфат амонію буває ІІІ сортів і містить азот в перерахунку на суху речовину не менше 20,8%, вологи не більше 0,2-0,3%, і вільної сірчаної кислоти не більше 0,005%. Аміачна селітра випускається марки А і Б. Для сірчаної кислоти марки Б, що містить нітратний і аміачний азот не менше 34,2% і фосфати в перерахунку на P2O5 - 0,5%.
Визначення аміачного азоту.
Азот, який міститься в добриві амонію визначається методом відношення, або формальдегідним методом, який є простіший і швидший, бо визначення азоту проводять титруванням. Визначення азоту відгонкою потребує спеціального обладнання і затрат часу, але він є більш точним.
Формальдегідний метод.
Амонійні солі при взаємодії з формальдегідом, виділяють еквівалентну кількість кислоти, яку відтитровують розчином NaOH по фенолфталеїну:
2(NH4)2SO4 + 6HCOH = (CH2)6N4 + 2H2SO4 + 6H2O
4NH4NO3 + 6HCOH = (CH2)6N4 + HNO3 + 6H2O
4NH4Cl + 6HCOH = (CH2)6N4 + HCl + 6H2O
Вміст амонійного азоту обчислюють за формулою:
X = V*0.0014*250*100 / 25*g
V – об’єм NaOH 0,1 н розчину витраченого на титрування
g – наважка проби.
Метод відгонки аміаку
. Амонійні солі при взаємодії з лугом виділяють аміак, який поглинають титрованим розчином кислоти взятим у надлишку:
(NH4)2SO4 + 2NaOH = Na2SO4 + 2NH3 + 2H2O
2NH3 + H2SO4 = (NH4)2SO4
Надлишок кислоти визначають титруванням:
H2SO4 + 2NaOH = Na2SO4 + 2H2O
Процентний вміст аміаку вираховують за формулою:
X(NH3) = (V1*K1 – V2*K2)*0.00852*100 / g
g – наважка добрива
V1 – об’єм H2SO4 взятий для аналізу
V2 – об’єм NaOH, що пішов на титрування надлишку H2SO4.
Державний стандарт на аміачну селітру
1. Зовнішній вигляд – показник А гранульований продукт, без по сторонніх механічних домішок;
2. Стандартний вміст нітратного і аміачного азоту – 54%;
3. Вміст вологи метод сушки – 0,3%, методом Фішера – 0,6;
4. Вміст добавок в перерахунку на суху речовину: А – нітратів кальцію і магнію 0,2-0,5%, Б – фосфати 0,5-1,2%, В – сульфатів амонію 0,3-0,7;
5. рН 10% водного розчину не менше 4;
6. Вміст речовин нерозчинних в 10% розчині азотної кислоти не більше 0,2%;
7. Розчинність у % не менше 100;
8. Гранулометричний склад: вміст гранул 1-4 мм 93%, 2-3 мм не менше 50%, менше 1 мм не більше 5%;
9. Залинок на : 5 мл не повинен бути відсутній;
10. Механічна міцність гранул на роздавлюванні не менше 400 г на 1 ампулу.
Аналіз фосфатних добрив
До фосфатних добрив відносять супер фосфат простий (К(Н2РО4)*2Н2О+СаSО4), який містить Н-20%, Р2О5-13№, вологи і не більше 5% Н3РО4; подвійний супер фосфат – 41-52% Р2О5, томасшлак, преципітат, фосфорна мука Са3(РО4)2 – 19-30%Р2О5, апатити, фосфорити, кісткова мука Са3(РО4)2*СаСО3 – вони відносяться до незасвоєних фосфорних добрив.
Визначення Р
2
О
5
це визначення ваговим або фото колориметричним методами. Природний фосфорит – розчин в кількості осаджують магнезіальною сумішшю у вигляді MgNH4PO4, який після прокалювання переходить в Mg2P2O7, який зважують, засушують і проводять розрахунок в перерахунку на Р2О5:
3Ca3(PO4)2*CaF2 + 24HCl = 6H3PO4 + 12CaCl2 + 6HF
H3PO4 + MgCl2 + 3NH4OH = MgNH4PO4 + 3H2O + 2NH4Cl
2MgNH4PO4 = Mg2P2O7 + 2NH3 + H2O
P2O5 обчислюють вміст за формулою: x = g2*0.6377*250*100 / 50*g
g2 – маса осаду
g – маса наважки
0.6377 - коефіцієнт перерахунку маси Mg2P2O7 на масу Р2О5.
Фотоколориметричний метод визначення Р
2
О
5
метод оснований на взаємодії іонів фосфору і молібдену алюмінію з утворенням розчину синього кольору. Інтенсивність забарвлення пропорційна вмісту фосфорної кислоти в розчині. На фотоколориметрі визначають оптичну густину даного розчину, а по калібрувальній кривій обчислюють концентрацію Р2О5 % вміст за формулою:
x(Р2О5) = C*250*100 / 100*g
C – кількість Р2О5 знайдена по калібрувальному графіку
g – наважка фосфориту.
Оптичну густину визначають на ФЕК із зеленим світлофільтром із кюветною довжиною 10 мм. Перед тим будують калібрувальний графік по серії стандартних розчинів.
Аналіз калію в мінеральних добривах
В калійних мінеральних добривах визначають ваговим методом або методом полум’яної фотометрії.
Ваговий тетрафенілбуратний метод визначення калію в калійних добривах оснований на тому, що калій осаджують тетрафенілбуратом натрію в оцтово-кислому середовищі з подальшим висушуванням і зважуванням, одержують осад тетрафенілбурат калію. Осадження проводять на водяній бані до 40˚С. Осаду дають 5 хв відстоятися, охолоджують в ексикаторі з холодною водою, фільтрують через попередньо висушений і зважений фільтруючий тигль. Промивають три рази холодною водою, висушують в сушильній шафі при температурі 120˚С до постійної маси. Проводять розрахунки, за результати аналізу приймають середнє арифметичне значення двох паралельних значень, розходження між якими не перевищує 0,3%, коефіцієнт перерахунку К=20 на КCl складає 1,583.
Полумяно фотометричний метод визначення калію в складних мінеральних добривах
Метод оснований на вимірюванні інтенсивності вилучення калію, яке вводиться в полум’я у вигляді аерозолю. В полум’яній фотометрії використовують ПФН, ПФА-1 чи Сатурн. Будують калібрувальний графік, вимірюють інтенсивність випромінення стандартних розчинів, відкладаючи на осі абсцис концентрацію, а на осі ординат відповідні покази приладу.
Хід аналізу
50 мл розчину добрива відфільтровують, відбираючи піпеткою в мірну колбу на 250 мл, доводять до мітки, перемішують. 25 мл одержаного розчину, що відбирають в мірну колбу на 250 мл, доводять до мітки і перемішують. Одержаний розчин вводять в полум’яний фотометр, знімаючи показники приладу і по графіку знаходять концентрацію калію в аналізую чому розчині. Вимірюють з повтором, одержують другий результат. За результат аналізу приймають середнє арифметичне значення двох визначень, розподіл між якими не перевищує 5%. Масову долю калію в перерахунку на калій – 20, вираховують за формулою:
X = C1*C2 / 2*(100*500*200*1.205 / 5*n) %
1.205 – коефіцієнт перерахунку К2О
n – маса наважки аналізуючої проби
C1 – C2 – концентрація калію одержаного по калібрувальному графіку.
|
