ЛЕКЦИЯ
ПРОГРАММА “ГЕНОМ ЧЕЛОВЕКА”
Геном секвенировали в 2003 г‚ т.е. к пятидесятилетнему юбилею открытия двойной спирали ДНК (1953)‚ планировалось к 2005 г.
В 1988 г. один из первооткрывателей знаменитой двойной спирали ДНК, нобелевский лауреат Дж. Уотсон, публично высказал мысль о том, что наука вплотную приблизилась к раскрытию химической основы наследственности, причем не какого-либо низшего организма, а "царя природы" - человека. В том же самом 1988-м с аналогичной идеей выступил выдающийся российский молекулярный биолог и биохимик, академик А.А. Баев (1904-1994). После консультаций с коллегами он обратился к М.С. Горбачеву с письмом, в котором предложил организовать государственный научный проект по изучению генома человека. В России, как и за ее пределами, эта идея также была встречена весьма критически, однако время шло, и очень скоро научное сообщество во всем мире стало обсуждать ее всерьез. С 1989 г. и в США, и в СССР функционируют соответствующие научные программы; позднее в 1999 г. возникла Международная организация по изучению генома человека (HUGO‚ HumanGenomeProject), вице-президентом которой несколько лет был академик А.Д. Мирзабеков. Это один из самых дерзновенных, дорогостоящих и потенциально важных проектов в истории цивилизации. Если в 1990 г. на него было потрачено около 60 млн долларов в целом, то в 1998 г. одно только правительство США израсходовало 253 млн долларов, а частные компании – и того больше.
Координационный центр HUGO находится в американском городе Бетесда, недалеко от Вашингтона, и относится к системе национальных институтов здоровья (National Institutes of Health). Возглавляет его Фрэнсис Коллинз - директор Института геномных исследований в Бетесде. Центр координировал научную работу в шести странах - Германии, Англии, Франции, Японии, Китае и США. Но национальные программы по геномике сегодня имеют более 20 стран (20 лабораторий), а членами HUGO являются представители более 50 стран. Национальные программы есть в развивающихся странах, например в Китае и Бразилии, где правительства понимают важность геномной программы. В научном совете много лет работали А. Мирзабеков и я. Сейчас Россию в нем представляет профессор Н. К. Янковский.
Реклама

Важно подчеркнуть, что с самого начала работ по геномному проекту мир договорился об открытости, доступности всей получаемой информации для его участников независимо от их вклада и государственной принадлежности. Это значит, что любая лаборатория, закончив расшифровку нуклеотидной последовательности какого-либо фрагмента ДНК, немедленно посылает результаты в международную базу данных в Америку или Германию. Из таких баз данных ученые, занимающиеся биоинформатикой, черпают информацию для своих расчетов. Сейчас существуют десятки мощных баз данных, в которых аккумулирована гигантская информация о структуре не только генома человека, но и геномов многих других организмов.
В 1989 г в СССР по решению правительства было открыто финансирование и организован Научный совет по программе "Геном человека" под руководством А.А. Баева. Расположившийся в головном учреждении программы - Институте молекулярной биологии им. В.А. Энгельгардта РАН, совет весьма быстро создал инфраструктуру, объединил исследования многих разрозненных групп. В России по проекту работает около 100 групп. Все хромосомы человека поделены между странами-участницами, и России для исследования достались 3-, 13- и 19-я хромосомы.
Российская программа развивалась по ряду направлений: медицинская геномика‚ функциональная геномика и
биоинформатика.
Одно из главных - биоинформатика. Что это такое? Биоинформатика - компьютерный анализ всей совокупности данных по нуклеотидным последовательностям ДНК. Сейчас в базах данных находится несколько миллиардов нуклеотидных пар человеческого генома и геномов других живых организмов. В этом море информации еще нужно разобраться, описать, понять, что следует за чем, где начало гена, где его конец, где регуляторные участки. Не определить, а предсказать. Расшифровать нуклеотидную последовательность - это все равно, что читать книгу, просто произнося названия букв подряд. Найти ген, значит понять, как буквы складываются в слова. Вероятность правильного предсказания сегодня достигает 85%. Биоинформатика не дает конечной информации, она дает исходную информацию. А затем наличие того или иного гена проверяется экспериментально. Биоинформатика предсказывает: вот здесь ген начинается, а здесь - заканчивается. Ученые-экспериментаторы "вырезают" предполагаемый ген из ДНК и проверяют, действительно ли этот фрагмент отвечает за синтез определенной белковой молекулы. Иногда оказывается, что ученые-биоинформатики предсказали гены правильно, а иногда - нет.
Реклама

Еще один аспект, который особенно бурно развивается в многонациональной России – это определение генома разных народностей
. Ее населяют разные этнические группы. Оказывается, что геном у разных народностей слегка различается. Можно в ДНК выделить определенный "рисунок" нуклеотидов (особое расположение), который будет говорить о том, что этот человек - башкир, а этот - татарин. Геномы представителей разных этнических групп не идентичны, но различия между ними чрезвычайно незначительны, хотя и абсолютно достоверны, и поэтому возможно сравнивать разные этнические группы.
Такой подход связывает геномику с историей, лингвистикой, археологией, палеонтоло гией, этнографией. И возникают поразительно интересные находки. Как вы думаете, к какой этнической группе ближе всего русские?
Славяне близки по материнской линии (поскольку изучается митохондриальная ДНК, передающаяся ребенку от матери) к нашим западным соседям: немцам, угрофиннам.
Сейчас ведутся работы по изучению Y-хромосомы, что гораздо сложнее, чем изучать митохондриальную ДНК. Через два-три года мы будем знать, как выглядит русский этнос по отношению к своим соседям уже по отцовской линии.
Работа очень увлекательная и ведется весьма активно. Участвуют исследователи из Томска, Москвы, Уфы и Тарту (Эстония). Международное сообщество смотрит на результаты наших исследований во все глаза: ведь мы имеем уникальные этносы.
Прежде всего, можно будет подбирать лекарственные препараты "по национальному признаку". Ведь не секрет, что многие признаки сцеплены с принадлежностью человека к определенной этнической группе. Поэтому такие исследования не только очень интересны, но они еще и создают основу будущей индивидуальной медицины.
Новая медицина станет не только индивидуальной, а профилактической (превентивной). Врачи смогут не только лечить болезнь, но и предотвращать ее возникновение. Геномика позволит сделать и это
В ядре каждой соматической клетки человека содержится 23 пары хромосом: на каждую хромосому приходится по одной молекуле ДНК. Длина всех 46 молекул ДНК в одной клетке человека равна почти 2 м, количество нуклеотидных пар составляет 6,4 млрд. Общая длина ДНК во всех клетках человеческого тела (их примерно 5х1013
) составляет 1011
км, что почти в тысячу раз больше расстояния от Земли до Солнца. Число генов у человека находится в пределах от 30 до 40 тысяч.
Предположение о числе генов в геноме человека с момента начала проекта сократилось вдвое (от 80-100 тыс). Выявлено большое количество "бессмысленных" участков. Они устроены не так, как гены‚ более того – это значительная часть генома. (95%). Вот в 5% генома, где находятся те самые 32 тысячи генов, мы знаем многое о структуре и немногое о функциях. По-научному "бессмысленные" участки называют некодирующими. Некоторые американские ученые называют их "junk" - барахлом, мусором или "эгоистической ДНК". Однако‚ если мы не понимаем, для чего нужны какие-то участки ДНК, сие еще не значит, что они - мусор.
У бактерии "бессмысленных" участков вообще нет. У дрожжей почти нет. По мере повышения уровня организации живого организма накапливается все больше некодирующей ДНК. Я думаю, что некодирующие последовательности ДНК могут оказаться резервуаром эволюции, складом "запчастей". Если с каким-либо геном что-то не в порядке, возможно, клетка использует фрагменты некодирующей ДНК для ремонта поврежденного гена.
В "бессмысленной" ДНК есть испорченные гены, погибшие в результате каких-то мутаций. Их называют псевдогенами.
Во-вторых, наши далекие предки - неандертальцы или кроманьонцы болели вирусными заболеваниями, и эти вирусы (а вирусы состоят из молекул ДНК или РНК и белковой оболочки) иногда попадали в геном и оставались там навсегда. Иными словами, часть нашего генома - молекулярное кладбище древнейших вирусов.
Затем, в нашем геноме есть масса повторяющихся участков. Действительно, очень интересно, почему человек - "венец эволюции" имеет огромную долю "неработающего" генома.
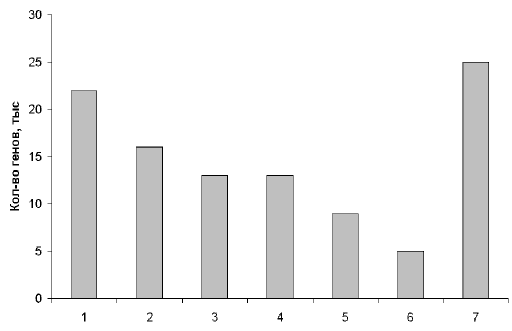
Рис. 2. Примерное распределение генов человека по их функциям.
1 – производство клеточных материалов; 2 – производство энергии и ее использование; 3 – коммуникации внутри и вне клеток; 4 – защита клеток от инфекций и повреждений; 5 – клеточные структуры и движение; 6 – воспроизводство клеток; 7 – функции не выяснены
По своему геному мы мало отличаемся от мыши. Различия в структуре генов - процентов 10-15, не больше. А от шимпанзе мы отличаемся на 1‚23%. Это показало первое в мире исследование, проведенное международной группой специалистов во главе с японским профессором Иосиюки Сакаи.
Проблема происхождения человека стала гораздо сложнее, чем ученые думали раньше. Подсознательно мы надеялись набрать сотню генов, отличающих человека от шимпанзе. И мы скажем по-французски "voila" - вот они эти гены, благодаря которым мы "выбились" в люди. А пока их нет.
Различия обнаружены в другом: в геноме человека много вставленных в него чужеродных элементов – ретровирусов, а у обезьян их почти нет.
Основной задачей программы является построение исчерпывающих генетических карт большого разрешения каждой из хромосом человека, которое должно завершиться определением полной первичной структуры ДНК всех хромосом.
В течение последних лет исследования проводились в следующих направлениях:
1. Компьютерный анализ полного генома человека и его частей на основе информации в открытых базах данных. Разработка принципиально новых подходов к хранению, обработке и получению структурной информации из баз данных на основе вновь созданного программного обеспечения.
2. Идентификация новых генов на основе физического, хромосомного и функционалного картирования, клонирования и секвенирования. Структурный и функциоеналный анализ вновь найденных генов и регуляции их активности.
3. Установление cause-and-effect генетических отношений между генами и предрасположенностью к широкораспространенным заболеваниям различной природы. Выявление роли индивидуальных генов и их мутаций в этиологии и развитии некоторых заболеваний человека.
4. Развитие методов генной и геномной диагностики заболеваний человека на основезнания физической карты и последовательностей нуклеотидов.
5. Разработка методов генной терапии моногенных заболеваний на основе знаний о молекулярно-генетических механизмах их возникновения и развития.
6. Разработка открытых юридических, этичских, законодательных/ правовых, социальных и других аспектов исследований генома и использзования информации о структуре и свойствах геномов отдельных ??? людей. Предсказания путей развития медицины и здравоохранения на основе нового уровня знаний о геноме человека и формулирование соответствующих практических предложений.
Решение основной задачи программы «Геном человека» включает следующие этапы.
• На первом этапе необходимо завершить составление детальной генетической карты
и отметить гены, отстоящие друг от друга на расстоянии, не превышающем в среднем 2 млн оснований (1 млн оснований равен 1 мегабазе — 1 Мб, от англ. base—
основание).
• Второй этап предполагает составление физических карт низкого разрешения каждой хромосомы (разрешение 0,1 Мб).
• На третьем этапе следует получить физическую карту высокого разрешения всего генома в виде охарактеризованных по отдельности клонов (клон содержит 5 Кб).
• Четвертый этап посвящен определению полной первичной структуры (секвенированию) всей ДНК генома человека (разрешение — 1 основание).
• На пятом, заключительном, этапе необходимо в найденных последовательностях нуклеотидов локализовать все гены организма и определить их функциональное значение.
Генетическое картирование
Генетические карты сцепления.
Генетические карты сцепления определяют хромосомную принадлежность и взаимное расположение генетических маркеров относительно друг друга. Картирование в узком смысле — определение положения гена или мутации в хромосоме. Позднее этот термин получил более широкое толкование. Он относится не только к гену, но к любому маркеру,
под которым подразумевают ген, мутацию, участок ДНК с неопределенной функцией, точку расщепления ДНК рестриктазами. Таким образом, маркер — это любой наследуемый признак, доступный идентификации тем или иным способом. Установление локализации какого-либо маркера позволяет использовать его для определения положения другого маркера.
На практике именно генетические карты сцепления и только они позволяют локализовать сложные генетические маркеры (например, ассоциированные с симптомами заболевания) на первых этапах исследования и дают возможность их дальнейшего изучения.
До начала 70-х годов XX в. построение генетических карт человека продвигалось очень медленными темпами. Первый ген человека (ген цветной слепоты) был картирован на Х-хромосоме в 1911 г., а первый аутосомный ген — только в 1968 г. К 1973 г. на хромосомах человека было картировано 64 гена, а к 1994 г. — 5000 структурных генов и свыше 60 000 маркерных ДНК-последовательностей. Столь стремительный прогресс в картировании генов человека связан с появлением новых технологий в цитогенети-ке, в клеточных культурах и особенно в молекулярной генетике.
Гибридизация соматических клеток.
Одним из наиболее популярных методов отнесения генетического маркера (функционально активного гена) к конкретной группе сцепления является гибридизация (слияние друг с другом) соматических клеток разных биологических видов организмов, один из которых — исследуемый. Гибридные клоны получают путем искусственного слияния клеток человека и различных грызунов: китайского хомячка, мыши, крысы. Культивирование таких соматических гибридов, как оказалось, сопровождается утратой хромосом человека. Потеря хромосом носит случайный характер, и образующиеся клоны клеток содержат оставшиеся хромосомы в разных сочетаниях. Так получают панели гибридных клеточных клонов, содержащих всего одну или несколько хромосом человека и полный набор хромосом другого вида. Обнаружение человеческих белков, специфических мРНК или последовательностей ДНК в таких клонах позволяет однозначно определить хромосомную принадлежность соответствующих генов.
Гибридизация
in
situ
(в том же месте).
Этот метод дает возможность локализовать определенные последовательности нуклеотидов на хромосомах. Они выступают в качестве зондов. Препараты фиксированных хромосом гибридизуют с исследуемыми последовательностями, меченными радиоактивной или флуоресцентной меткой. Меченые молекулы оказываются ассоциированными с участками хромосом, содержащими последовательности, комплементарные меченому зонду. Полученные гибриды анализируют с помощью микроскопа либо непосредственно, либо после радиоавтографии. Этот метод по частоте использования в последнее время прочно выходит на первое место. Наиболее популярной оказалась группа методов, получивших название флуоресцентной гибридизации
in
situ
—
метод FISH (от англ. Fluorescencein
situ
hybridization
).
Полимеразная цепная реакция (ПЦР) позволила быстро и эффективно амплифицировать почти любой участок генома человека, а полученные продукты ПЦР использовать в качестве зондов для картирования соответствующих участков на хромосомах путем гибридизации in
situ
. В этом плане успешно разработана концепция сайтов, привязанных к последовательностям, —STS(от англ. Sequence
-
tagged
sites
).
Все фрагменты ДНК, которые используются для построения генетических и физических карт, можно однозначно идентифицировать с помощью последовательности нуклеотидов длиной в 200 — 500 н.п., которая является уникальной для данного фрагмента. Эти сайты амплифицируют с помощью ПЦР и применяют в качестве зондов. STS позволили создать основу для разработки единого языка, дающего возможность разным лабораториям описать свои клоны. Конечным результатом разработки концепции STS является создание исчерпывающей карты STS генома человека. Для получения маркеров в настоящее время часто применяют праймеры, соответствующие диспергированным повторяющимся последовательностям, среди которых первыми стали использовать А1u-последовательности, так как они характерны именно для генома человека. Поскольку в геноме человека больше 90 % умеренно повторяющихся последовательностей представлены семействами А1u и КрnI (последние повторяются реже и обладают характерной локализацией в хромосомах), они и используются для получения соответствующих зондов в ПЦР-реакции.
Физические карты низкого разрешения.
Физические карты генома отражают реальное расстояние между маркерами, выражаемое в парах нуклеотидов. Физическую карту низкого разрешения часто называют хромосомной (цитогенетической) картой генома.
В начале 70-х годов XX в. появилась реальная возможность точной идентификации не только всех хромосом в кариотипе человека, но и их отдельных сегментов. Это связано с появлением мето да дифференциального окрашивания препаратов метафазных хромосом. Хромосомные препараты окрашивают некоторыми флуорохромами после соответствующей протеолитической обработки или нагревания. При этом на хромосомах выявляется характерная поперечная исчерченность — так называемые диски (бэнды), расположение которых специфично для каждой хромосомы. Величина небольших дисков на прометафазных хромосомах соответствует примерно 1 млн н.п. на физических картах. Каждая хромосома после дифференциальной окраски может быть разделена на сегменты, нумерация которых начинается от центромерного района вверх (короткое плечо р)
либо вниз (длинное плечо — q
)
. Полосы в каждом сегменте также пронумерованы в аналогичном порядке. Запись положения гена на карте включает номер хромосомы, плечо, номер сегмента, бэнда и его субъединицы.
Запись 7 q21.1 означает, что ген локализован в субъединице 1-го бэнда 2-го сегмента длинного плеча хромосомы 7. Подобная запись удобна для цитогенетического картирования метода гибридизации insitu, позволяющего локализовать ген с точностью до одного бэнда и даже его субъединицы.
Хромосомные карты генома человека получают также локализацией генетических маркеров, чаще всего методом FISН: для метафазных хромосом разрешающая способность хромосомных карт находится в пределах 2 — 5 млн н.п.; для интерфазных хромосом (генетический материал находится в менее компактной форме) — приближается к 100 тыс. н.п. Для этого уровня картирования характерны карты кДНК (с.
358). Они отражают положение экспрес-сирующихся участков ДНК (экзонов) относительно известных ци-тогенетических маркеров (бэндов) на метафазных хромосомах. Поскольку такие карты дают представление о локализации транскрибирующихся участков генома, в том числе и генов с неизвестными функциями, они могут быть использованы для поиска новых генов. Этот подход полезен при поиске генов, повреждение которых вызывает заболевания человека, в том случае, если приблизительная локализация таких участков хромосом уже проведена на генетических картах сцепления (см. рис. 100).
Физические карты высокого разрешения.
Для построения физических карт высокого разрешения экспериментально реализуется два альтернативных подхода: картирование сверху вниз и картирование снизу вверх (рис.В к геному)
. Для картирования сверху вниз препарат ДНК индивидуальной хромосомы человека разрезают крупнощепящими рестриктазами (например, NotI) на длинные фрагменты, которые после разделения методом электрофореза в пульсирующем поле подвергаются дальнейшей обработке другими рестриктазами.
Методом электрофореза под действием однонаправленного постоянного поля в агарозном или полиакриламидном гелях удается разделить фрагменты ДНК размером не более 30 —50 тыс. н.п. Продвижение больших фрагментов ДНК в геле при пульсирующем изменении направления электрического поля происходит за счет конформационных изменений, обусловленных скручиванием и раскручиванием молекул ДНК в момент переключения направления поля. В этом случае удается разделить молекулы ДНК размером от 50 тыс. н.п. до 10 млн н.п.).
В результате получают макрорестрикционную
карту. Метод электрофореза был с успехом использован для картирования малых геномов.
Для картирования генома человека снизу вверх на основе препарата суммарной ДНК генома или индивидуальной хромосомы получают серию случайных клонов протяженных последовательностей ДНК (10— 1000 тыс. н.п.), часть из которых перекрывается друг с другом. В качестве вектора для клонирования в этом случае используют искусственные минихромосомы дрожжей (УАС). Последовательный набор клонов, содержащих частично перекрывающиеся и дополняющие друг друга фрагменты ДНК из определенного района генома, получил название скользящего зондирования,
или «прогулки по хромосоме». Каждый раз отобранный фрагмент используется в качестве ДНК-зонда для последующего поиска. В результате получают набор клонированных фрагментов ДНК, полностью перекрывающих исследуемый участок генома, получивший название «контиг». Эта стратегия впервые была успешно применена для изучения 3-й хромосомы дрозофилы. С ее помощью редко удается пройти более 200 — 300 тыс. н.п. в одном направлении из-за наличия в геноме повторяющихся и трудно клонируемых последовательностей ДНК. Для преодоления таких ограничений и ускорения процесса поиска генных последовательностей Ф. Коллинз, ныне президент Международного консорциума, предложил метод «прыжков» по хромосоме, позволяющий изолировать фрагменты ДНК, отстоящие в геноме друг от друга на сотни тысяч пар нуклеотидов (длина прыжка), не выделяя при этом все промежуточные последовательности ДНК.
Правильность полученных контигов подтверждают обычно гибридизацией insitu (FISH) с одновременной привязкой к определенным участкам исследуемых хромосом.
Определение нуклеотидной последовательности генома человека
Исчерпывающая физическая карта генома человека должна представлять собой полную последовательность нуклеотидов ДНК всех его хромосом. К решению такой грандиозной по объему задачи привлечены многие хорошо финансируемые лаборатории в разных странах мира, оснащенные автоматическими высокопроизводительными секвенаторами.
Создание в середине 70-х годов теперь уже прошлого века двух различных методов расшифровки нуклеотидной последовательности ДНК. Хронологически первым был метод Максама - Гилберта. В его разработке большую роль сыграл академик Андрей Дарьевич Мирзабеков. Английский ученый Фред Сэнгер предложил другой способ расшифровки структуры ДНК. За разработку этих методов Гилберт и Сэнгер получили Нобелевскую премию. Интересно, что для Сэнгера эта премия уже вторая, первую он получил за расшифровку аминокислотной последовательности белка инсулина. Случай в науке уникальный - один и тот же человек первым расшифровал структуру и белка и ДНК!
- Метод Максама - Гилберта состоит в том, что молекулу ДНК разбивают на кусочки, затем эти кусочки подвергают химическим воздействиям, потом специальным образом обрабатывают. Ученые смотрят, что при этом происходит с нуклеотидной последовательностью, и на основании этого делают вывод о порядке расположения нуклеотидов друг за другом в каждом фрагменте ДНК.
Согласно методу Сэнгера молекулу ДНК с помощью специальной обработки ферментами не только расщепляют на фрагменты, но и "расплетают" ее двойную спираль на две нити. Потом по каждому из полученных обрывков, состоящих из отдельных нуклеотидных "нитей", с помощью специальных химических "затравок" восстанавливается недостающая вторая нить нуклеотидов. Но не полностью - ее синтез обрывают на разных нуклеотидах. При этом получался набор цепей ДНК с непрерывно изменяющейся длиной - "лесенка". Фрагменты разной длины помечены на концах флуоресцентной меткой, чтобы их было легко обнаружить.
Надо сказать, что российские биологи внесли существенный вклад в разработку и этого метода. Новосибирский ученый профессор Станислав Константинович Василенко предлагал принцип "лесенки" еще до публикации работ Сэнгера, этот же принцип развивал и академик Евгений Давыдович Свердлов, директор Института молекулярной генетики РАН. То, что Василенко и Свердлов - предтечи Сэнгеровского метода, забывать не стоит.
Все автоматы-секвенаторы построены по принципу метода Сэнгера, поскольку он оказался более удобным для автоматизации и комьютерной регистрации. Выпущено огромное количество автоматов и стандартных наборов реактивов для анализа. По сути, секвениро вание (то есть определение нуклеотидной последовательности ДНК) стало рутинной лаборантской работой. А метод Максама-Гилбера имеет скорее историческое, чем практическое значение.
Еще 15-20 лет назад расшифровка нуклеотидной последовательности в 1000 нуклеотидов считалась почти научным подвигом, за это можно было сразу получить степень доктора наук. Но уже к 1990 году секвенирование ДНК стало массовой технологией. А сейчас квалифицированный лаборант проделывает такую работу меньше, чем за один день.
Разработаны и другие совершенно новые методы секвенирования. Один из них базируется на возможности избирательно присоединять тяжелые атомы металлов (нерадиоактивные изотопы) к определенным нуклеотидам с последующим масс-спектрометрическим сканированием молекул ДНК, пропускаемых через тончайший (нанометровый) микрокапилляр. Устройство читает нуклеотидную последовательность практически безошибочно. При этом не нужно дорогостоящих и отнимающих уйму труда операций по химическому секвенированию, использованию наборов рестрикционных ферментов и прочих ухищрений. Метод начала использовать компания "Секвеном", зарегистрированная в городе Сан-Диего (Калифорния) и руководимая Чарлзом Кэнтором.
Другой подход основан на присоединении флюоресцентных меток к ДНК, разрезании ДНК одним или несколькими рестрикционными ферментами на достаточно протяженные куски и оптическом анализе кусков. Так как флюоресцентные метки, сорбирующиеся на индивидуальных нуклеотидах, создают для каждого участка ДНК светящуюся картинку, характерную только для него, можно сравнивать ее с имеющимися в памяти компьютеров картинками. Для этого сотрудники компании "Силера джиномикс" создали прибор оптического "обстрела" протяженных ДНК (система Visionade) и математический алгоритм Gentig. Если после оптического просмотра остаются сомнения в точности нуклеотидных последовательностей в каких-то коротких участках, только эти участки и надлежит секвенировать химически. Оптическая "стрельба" по нарезанным участкам ДНК позволила достичь небывалой скорости в секвенировании: в свое время изучение генома кишечной палочки потребовало работы нескольких сот человек в течение 12 месяцев, в то время как система Visionade помогла расшифровать этот же геном в несколько минут.
Структура генома человека (по данным секвенирования на 2001 г.)
На основе компьютерных алгоритмов, построенных на современных представлениях об общей структуре гена и о белковых доменах, было рассчитано количество генов, кодирующих белки в геноме человека. Международный консорциум определил 31 780 белок-кодирующих генов, а фирма Целера Геномикс обнаружила 39 114 таких генов.
Показано, что типичный ген человека состоит примерно из 28000 н.п. и имеет 8 экзонов, его кодирующая последовательность 1340 н.п., этот ген кодирует 447 аминокислот.
Самым большим геном, найденным в геноме человека, является ген мышечного белка дистрофина (2,4 • 106
н. п.). Фибриллярный белок титин, ответственный за пассивную эластичность скелетных мышц, состоит из 27 000 аминокислотных остатков. Его ген содержит 234 экзона. Это наибольшее количество экзонов, пока найденное в белок-кодирующих генах человека. Структура и организация генов человека много сложнее, чем структура генов других эукариот. Очень часто они прерываются большими интронами, 35 % генов человека могут считываться с разных рамок, а 40 % РНК подвергаются альтернативному сплайсингу. Таким образом, одна последовательность ДНК может кодировать более одного вида мРНК.
По сравнению с геномами других эукариотических организмов у человека большее распространение получили гены, участвующие в обеспечении иммунной защиты; в развитии нервной системы (нейротрофические факторы, факторы роста нервов), сигнальных молекул, миелиновых белков, потенциал-управляемых ионных каналов и синаптических рецепторных белков; в построении цитоскелета и движении везикул, обеспечении внутри- и межклеточной сигнализации, поддержании гомеостаза. У человека значительно большее количество генов участвует в транскрипции и трансляции. Из 2000 таких генов 900 относятся к семейству белков, содержащих «цинковые пальцы».
В целом на долю генов, кодирующих белки, приходится 2 %
генома; на области, кодирующие РНК, — около 20% генома, повторяющиеся последовательности занимают более 50 % генома, причем значительная часть этой ДНК возникла за счет обратной транскрипции РНК.
Исследование структуры генома ряда прокариот и эукариот, и человека в частности, способствовало созданию науки о геномах — геномики.
В нее включают изучение геномов на молекулярном, хромосомном, биохимическом и фенотипическом уровнях. Нам представлена схема, поясняющая взаимоотношения между геномикой человека и другими научными направлениями в современной биологии. Структурная и сравнительная геномика через биоинформатику переходит в новый раздел — функциональную геномику, главной задачей которой является выяснение биологических функций генных продуктов и в первую очередь белков.
У многих современных исследователей, работающих в области геномики, нет сомнений, что первое десятилетие XXI в. будет эрой функциональной геномики и биоинформатики.
В сети Интернет можно найти большое число адресов, содержащих разнообразную информацию, касающуюся генома человека:
Что можно ждать от геномных исследований в ближайшие 40 лет? Вот как сформулировал прогноз Ф.Коллинз, руководитель программы "Геном человека" (США).
2010 год
Генетическое тестирование, профилактические меры, снижающие риск заболеваний, и генная терапия до 25 наследственных заболеваний. Медсестры начинают выполнять медико-генетические процедуры.
Широко доступна преимплантационная диагностика, яростно обсуждаются ограничения в применении данного метода. В США приняты законы для предотвращения генетической дискриминации и соблюдения конфиденциальности. Не всем доступны практические приложения геномики, особенно в развивающихся странах.
2020 год
На рынке появляются лекарства от диабета, гипертонии и других заболеваний, разработанные на основе геномной информации. Терапия рака, прицельно направленная на свойства раковых клеток. Фармакогеномика становится общепринятым подходом для создания многих лекарств. Изменение способа диагностики психических заболеваний, появление новых способов их лечения, изменение отношения общества к таким заболеваниям. Демонстрация безопасности генотерапии на уровне зародышевых клеток при помощи технологии гомологичной рекомбинации.
2030 год
Определение последовательности нуклеотидов всего генома отдельного индивида станет обычной процедурой, стоимость которой менее 1000 $. Каталогизированы гены, участвующие в процессе старения. Проводятся клинические испытания по увеличению максимальной продолжительности жизни человека. Лабораторные эксперименты на человеческих клетках заменены экспериментами на компьютерных моделях. Активизируются массовые движения противников передовых технологий в США и других странах.
2040 год
Все общепринятые меры здравоохранения основаны на геномике. Определяется предрасположенность к большинству заболеваний (при/до рождения).
Доступна эффективная профилактическая медицина с учетом особенностей индивида. Болезни детектируются на ранних стадиях путем молекулярного мониторинга.
Для большинства заболеваний доступна генная терапия. Замена лекарств продуктами генов, вырабатываемыми организмом при ответе на терапию. Средняя продолжительность жизни достигнет 90 лет благодаря социоэкономическим мерам. Проходят серьезные дебаты о возможности человека контролировать собственную эволюцию.
Протеомика
Эта совершенно новая отрасль биологии, изучающая структуру и функции белков и взаимосвязи между ними, названа по аналогии с геномикой, занимавшейся геномом человека. Само рождение протеомики уже объясняет, зачем нужна была программа Геном человека. Поясним на примере перспективы нового направления
Вернемся к протеомике. Знание аминокислотных последовательностей и трехмерной структуры определенных белков позволило разработать программы сопоставления генетических последовательностей с аминокислотными, а затем программы предположительного расположения их в трехмерной структуре полипептидов. Знание трехмерной структуры позволяет быстро находить химические варианты молекул, в которых блокирован, например, активный центр, или определять положение активного центра у мутантного фермента.
|
