Фізичні властивості молібдену
Молібден (лат. Molybdaenum), Mo, хімічний елемент VI групи періодичної системи Менделєєва; атомний номер 42, атомна маса 95,94; світло-сірий тугоплавкий метал. У природі елемент представлений сімома стабільними ізотопами з масовими числами 92, 94-98 і 100, з яких найбільш поширений 98
Мо (23,75%). Аж до 18 століття основний мінерал молібден молібденовий блиск (молібденіт) не відрізняли від графіту і свинцевого блиску, оскільки вони дуже схожі на вигляд. Ці мінерали носили загальну назву "молібден" (від греч. molybdos - свинець).
Елемент молібден відкрив в 1778 році шведський хімік К. Шееле, що виділив при обробці молібденіту азотною кислотою молібденову кислоту. Шведський хімік П. Гьельм в 1782 році вперше одержав металевий молібден відновленням МоО3
вуглецем.
Розповсюдження молібдену в природі. Молібден - типовий рідкісний елемент, його вміст в земній корі 1,1·10-4
%(по масі). Загальне число мінералів молібдена 15, велика частина їх (різні молібдати) утворюється в біосфері. У магматичних процесах молібден пов'язаний переважно з кислою магмою, з гранітоїдамі. У мантії молібдену мало, в ультраосновных породах лише 2·10-5
%. Накопичення молібдену пов'язане з глибинними гарячими водами, з яких він осідає у формі молібденіту MoS2
(головний промисловий мінерал молібдену), утворюючи гидротермальниє родовища.
Геохімія молібдену в біосфері тісно пов'язана з живою речовиною і продуктами його розпаду; середній вміст молібдену в організмах 1·10-5
%. На земній поверхні, особливо в лужних умовах, Mo (IV) легко окислюється до молібдатов, багато з яких порівняльне розчинні. У ландшафтах сухого клімату молібден легко мігрує, накопичуючись при випаровуванні в соляних озерах (до 1·10-3
%) і солончаках. У вологому кліматі, в кислих ґрунтах молібден часто малорухливий; тут потрібні добрива, що містять молібден (наприклад, для бобів).
У річкових водах молібдену мало (10-7
- 10-8
%). Поступаючи із стоком в океан, молібден частково накопичується в морській воді (в результаті її випаровування молібдену тут 1·10-6
%),частково осідає, концентруючись в глинистому мулі, багатому органічною речовиною і H2
S. Крім молібденових руд, джерелом молібдену служать також деякі мідні і мідно-свинцево-цинкові руди.
Реклама

Фізичні властивості молібдену. Молібден кристалізується в кубічну об'емоцентровану гратку з періодом а = 3,14Å. Атомний радіус 1,4 Å, іонні радіуси Мо4+
0,68 Å, Мо6
+ 0,62 Å. Густина 10,2 г/см3
(20 °С); tпл
2620 °С; tкип
близька до 4800 °С. Питома теплоємність при 20-100°С 0,272 кдж/(кг·К), тобто 0,065 кал/(г·град). Теплопровідність при 20°С 146,65 вт/(м·К), тобто 0,35 кал/(см·сек·град). Термічний коефіцієнт лінійного розширення (5,8-6,2)·10-6
при 25-700 °С. Питомий електричний опір 5,2·10-8
ом·м, тобто 5,2·10-6
ом·см; робота виходу електронів 4,37 ев. Молібден парамагнетик; атомна магнітна сприйнятливість -90·10-6
(20 °С).
Механічні властивості молібдену залежать від чистоти металу і попередньої механічної і термічної його обробки. Так, твердість по Брінеллю 1500-1600 Мн/м2
, тобто 150-160 кгс/мм2
(для спеченого штабіка), 2000-2300 Мн/м2
(для кованого прутка) і 1400-1850 Мн/м2
(для відпаленого дроту); межа міцності для відпаленого дроту при розтягуванні 800-1200 Мн/м2
. Модуль пружності молібдену 285-300 Гн/м2
. Мо більш пластичний, ніж W. Рекристаллізуючий відпал не приводить до крихкості металу.
Отримання молібдену. Основною сировиною для виробництва молібдену, його сплавів і з'єднань служать стандартні молібденітові концентрати, що містять 47-50% Мо, 28-32% S, 1-9% SiO2
і домішки інших елементів. Концентрат піддають окислювальному випаленню при 570-600 °С в багатоподовых печах або печах киплячого шару. Продукт випалювання - недогарок містить МоО3
, з домішками. Чисту МоО3
, необхідну для виробництва металевого молібдену, одержують з недогарка двома шляхами: 1) сублімацією при 950-1100 °С; 2) хімічним методом, який полягає в наступному: недогарок вилуджують аміачною водою, переводячи молібден в розчин; з розчину молібдата амонія (після очищення його від домішок Cu, Fe) виділяють полімолібдати амонія (головним чином парамолібдат 3(NH4
)2
O·7МоО3
·nН2
О) методом нейтралізації або випаровування з подальшою кристалізацією; прожаренням парамолібдата при 450-500 °С одержують чистий МоО3
, що містить не більш 0,05% домішок.
Металевий молібден одержують (спочатку у вигляді порошку) відновленням МоО3
в струмі сухого водню. Процес ведуть в трубчастих печах в дві стадії: перша - при 550-700 °С, друга - при 900-1000 °С. Молібденовий порошок перетворюють на компактний метал методом порошкової металургії або методом плавки. У першому випадку одержують порівняно невеликі заготовки (в перерізі 2-9 см2
при довжині 450-600 мм). Порошок молібдену пресують в сталевих прес-формах під тиском 200-300 Мн/м2
(2000-3000 кгс/см2
). Після попереднього спікання (при 1000-1200 °С) в атмосфері водню заготовки (штабіки) піддають високотемпературному спіканню при 2200-2400 °С. Спечений штабік обробляють тиском (кування, протяжка, прокатка). Крупніші спечені заготовки (100-200 кг) одержують при гідростатичному пресуванні в еластичних оболонках. Заготовки в 500-2000 кг виробляють дуговою плавкою в печах з охолоджуваним мідним тіглем і електродом, що витрачається, яким служить пакет спечених штабіков. Крім того, використовують електронопроменеву плавку молібдену. Для виробництва ферромолібдена (сплав; 55-70% Мо, інше Fe), призначеного для введення присадок молібдену в сталь, застосовують відновлення обпаленого молібденітового концентрату (недогарка) феросиліцієм у присутності залізної руди і сталевої стружки.
Реклама

Застосування молібдену. 70-80% молібдену, що видобувається, йде на виробництво легованих сталей. Решта застосовується у формі чистого металу і сплавів на його основі, сплавів з кольоровими і рідкісними металами, а також у вигляді хімічних з'єднань. Металевий молібден - найважливіший конструкційний матеріал у виробництві електроосвітлювальних ламп і електровакуумних приладів (радіолампи, генераторні лампи, рентгенівські трубки ); з молібдену виготовляють аноди, сітки, катоди, утримувачі нитки розжарення в електролампах. Молібденовий дріт і стрічка широко використовують в якості нагрівачів для високотемпературних печей.
Після освоєння виробництва великих заготовок. Молібден стали застосовувати (у чистому вигляді або з легуючими добавками інших металів) в тих випадках, коли необхідне збереження міцності при високих температурах, наприклад, для виготовлення деталей ракет і інших літальних апаратів. Для оберігання молібдену від окислення при високих температурах використовують покриття деталей силіцидом молібдену, жаростійкими емалями і іншими способами захисту. Молібден застосовують як конструкційний матеріал в енергетичних ядерних реакторах, оскільки він має порівняно малий перетин захоплення теплових нейтронів (2,6 барн). Важливу роль молібден виконує у складі жароміцних і кислототривких сплавів, де він поєднується головним чином з Ni, Co і Cr.
У техніці використовуються деякі з'єднання молібдену. Так, MoS2
- змащувальний матеріал для частин механізмів, що труться; дісиліцид молібдену застосовують при виготовленні нагрівачів для високотемпературних печей; Na2
MoO4
- у виробництві фарб і лаків; оксиди молібдену - каталізатори в хімічній і нафтовій промисловості.
Структурні методи дослідження речовини.
Рідкий стан речовини є проміжним між твердим і газоподібним (мал. 1.1). Область існування рідини обмежена з боку низьких температур переходом в твердий стан (точки cdd')
аз боку високих — переходом в газоподібний стан (точки с, е).ЛініяАК, щорозділяє рідку і газоподібну фази, закінчується критичною точкою, відповідній температурі Ткр
і
тиску Pкр
, вище за яких неможливе існування рідини в рівновазі з парою. Лінія рівноваги рідина — тверда фаза критичної точки не має. У металів температура плавлення підвищується із збільшенням тиску (крива АВ); у льоду, кремнію, германію — знижується (криваАВ').
ТочкаА надіаграмі стану відповідає температурі і тиску, при яких в закритій посудині знаходяться в рівновазі тверда, рідка і газоподібна фази. Рідині поєднують деякі властивості як твердих тіл, так і газів. Тверді тіла бувають кристалічні і аморфні. По типах зв'язку кристали діляться на атомні, іонні, металеві і молекулярні. Вони мають ближній і дальній порядок. Ближній порядок означає правильне розташування біля фіксованого атома, іона або молекули певного числа найближчих сусідів. Дальнім порядком називається розташування частинок в певній послідовності з утворенням єдиної тривимірної гратки. За наявності дальнього порядку відстань до будь-якого атома кристала обчислюється через параметри елементарної комірки по формулі
Ri =mi
a
+ni
b
+pi
c
(1)
де mi
ni
pi
координати атомів гратки.
Кристалічні тіла являються анізотропними, їх механічні, теплові, електричні і оптичні властивості у різних напрямах не однакові. Одна і та ж кристалічна речовина може знаходитися в декількох модифікаціях, які мають неоднакові структури. Так, вуглець існує у вигляді графіту, алмазу і карбіну, двоокис кремнію — у вигляді кварцу, тридиміта і крістабаліта;

сірка — у вигляді ромбічної і моноклінної модифікацій. Атоми, іони або молекули кристалів здійснюють узгоджені (колективні) коливання біля фіксованих положень рівноваги. У кристалах можливі пульсуючі рухи елементарних комірок, пульсації молекул, при яких вони періодично витягуються і стискаються у різних напрямах. Проведені І. Б. Берсукером дослідження показали, що пульсуючі рухи породжують поляризацію, роблять сильний вплив на оптичні, магнітні і інші властивості речовини. У газоподібному стані речовини атоми і молекули взаємодіють один з одним за допомогою ван-дер-ваальсових сил тяжіння на великих в порівнянні з розмірами частинок відстанях і квантово-механічних сил відштовхування на малих відстанях. Сили тяжіння в газах дуже слабкі, щоб надовго утримати молекули разом, тому розташування молекул в газі хаотичне. Молекули газу знаходяться в безперестанному русі, який відбувається не узгоджено (індивідуальних) у вигляді переміщень і зіткнень в кінці вільного пробігу. Кінетична енергія молекули газу значно більше потенційної. Якщо молекули багатоатомні, то при своєму поступальному русі вони можуть обертатися як ціле, здійснюють коливання, що становлять їх атоми.
Рідина—система динамічна. Атоми або молекули, зберігаючи ближній порядок у взаємному розташуванні, беруть участь в тепловому русі, який складніший, ніж в кристалі. Атоми і молекули рідини здійснюють коливання, як в кристалах, але положення рівноваги, щодо яких відбуваються ці коливання, не залишаються фіксованими. Зробивши деяке число коливань близько одного положення рівноваги, молекули переміщаються в сусіднє положення, обумовлюючи явище дифузії.
Переміщення молекул з одного рівноважного положення в інше може відбуватися: а) стрибком на відстань R1
близьке до середнього міжмолекулярного, і б) плавно разом з їх найближчим оточенням. У молекулах, що містять одинарні і подвійні зв'язки, можливі повороти атомів або груп атомів навколо цих зв'язків, що приводить до утворення ізомерів даного хімічного з'єднання. Згідне Я. І. Френкелю, тривалість перебування молекули в тимчасовому положенні рівноваги — час осілого життя — визначається по формулі:
 , (2) , (2)
де τ0
— період коливань молекули біля положення рівноваги; U— висота потенційного бар'єру, який відділяє один від одного два сусідні положення рівноваги; k— постійна Больцмана; T— термодинамічна температура. Числове значення τ залежить від будови і в'язкості рідини. По теоретичних розрахунках І. 3. Фішера, для аргону поблизу потрійної точки τ = 2,3 · 10-13
с; для води при кімнатній температурі τ = 10-10
с, τ0
= 1,4 · 10-12
с. Отже, кожна молекула води здійснює близько 100 коливань щодо одного і того ж положення рівноваги, перш ніж змінити місце.
По образному виразу Я.І. Френкеля, молекули в рідині ведуть в основному осілий спосіб життя, що є характерною межею рідкого стану, що зближує його з твердим тілом, з тією різницею, що в твердих тілах час осілого життя набагато більше, ніж в рідких. Із зростанням температури час осілого життя молекул в тимчасовому положенні рівноваги зменшується, відмінність між τ і τ0
поступово зникає. При цьому роль поступального руху молекул посилюється, а коливального — ослабляється, структура рідини все більш наближається до газової.
По теорії Я. І. Френкеля, у разі простих рідин через час осілого життя визначаються:
а) середня швидкість переміщення молекул рідини
 (3) (3)
б) самодифузія, що характеризує швидкість взаємного перемішування молекул,
 (3`) (3`)
в) в'язкість, що характеризує передачу імпульсу молекулами рідини,
 (4) (4)
Приведені формули відображають активаційний (прижковий) характер руху молекул. Досвід показує, що в рідинах молекули можуть переміщатися безперервно по траєкторіях дрейфу, без раптових стрибків. Такий тип руху домінує в зріджених інертних газах і в розплавлених металах. У асоційованих рідинах (наприклад, воді) більш вірогідний прижковий характер переміщення молекул.
Поступальне переміщення молекул рідини вкладає певний внесок в теплопровідність. Проте основним чинником, що визначає теплопровідність рідини, є процес розповсюдження пружних хвиль, породжених тепловими коливаннями молекул. Ці хвилі імітуються фононами (по аналогії з фотонами — квантами електромагнітних хвиль). Виходячи з кінетичної теорії теплопровідність для рідин можна виразити формулою:
 (5) (5)
де с — теплоємність рідини, v
— швидкість розповсюдження фононів, λ — довжина їх вільного пробігу. Ангармонізм коливального руху, безперервне переміщення молекул у всьому об'ємі рідини обмежує довжину вільного пробігу фононів, а отже, і теплопровідність.
У рідинах на відміну від газів домінують ті ж міжмолекулярні сили тяжіння, які обумовлюють той або інший тип зв'язку в кристалі. Так, наприклад, між атомами зріджених інертних газів діють ван-дер-ваальсовиє сили. Ті ж сили викликають взаємне тяжіння молекул неполярних рідин. Молекули води, кислот жирного ряду і спиртів взаємодіють один з одним за допомогою водневих зв'язків, виникнення яких пов'язане з наявністю в їх складі гідроксильних груп ОН. У розплавах солей діють електростатичні сили тяжіння, в металах — сили металевого зв'язку.
У рідкому германії, кремнії і інших напівпровідникових речовинах разом з металевим зв'язком частково зберігається ковалентний зв'язок. Кожна група рідин володіє специфічним ближнім порядком, успадковуваним від твердого тіла. Невелика відмінність густини рідин і кристалів, їх питомих теплоємностей і коефіцієнтів об'ємного розширення, з одного боку, указує на істотну відмінність теплот плавлення і паротворення, а з іншою — на те, що рідини по характеру взаємного розташування частинок, їх динаміці і взаємодії ближче до твердого, а не газового стану речовини. Я. І. Френкель писав, що зближення рідин з реальними газами допустиме лише у разі, коли рідина знаходиться при високих температурах, близьких до критичної, і володіє малою густиною. З другого боку, безперечним фактом є схожість їх з твердими тілами при температурах, близьких до температури кристалізації. Будучи фазою, проміжною між твердою і газоподібною, рідина, природно, знаходить безперервну гамму перехідних властивостей, примикаючи у області високих температур і великих питомих об'ємів до газів, а у області низьких температур і малих питомих об'ємів — до твердих тіл.
Особливості аморфного стану полягають у відсутності дальнього порядку і природної ізотропії властивостей. По структурі аморфні тіла нагадують рідини, а по характеру теплового руху — кристали. У аморфному стані можуть знаходитися як атомарні, так і молекулярні речовини.
Аналогія між структурами аморфних тіл і рідин не означає ідентичності існуючого в них ближнього порядку. Так, наприклад, рідкий: кремній і германій мають ближній порядок, істотно відмінний від ближнього порядку в аморфному стані. В той же час структура ланцюжка селену і теллура зберігається при переході з аморфного стану в рідкий. Різновидом аморфних тіл є стекла. Стеклоподібний стан речовини виходить з в'язкого розплаву при швидкому його охолоджуванні, тобто твердіє без кристалізації.
Кількісний опис рідини і аморфної речовини
В кристалічних тілах атоми, іони або молекули розташовані в певній послідовності, утворюючи тривимірну гратку. Вона складається з елементарних комірок, параметри яких можуть бути визначені експериментально по формулі Вульфа — Брегга:
2dsinи = nλ (6)
де θ— кут між площиною, що відображає, і падаючим пучком; d — міжплощіна відстань, пов'язана з трансляціями а, b, cкристалічної гратки і кутами α, β, γ між осями кристала. При температурі плавлення кристала його гратка руйнуються, зникає дальній порядок в розташуванні частинок, з'являється і складова трансляції теплового руху, унаслідок чого частково змінюється і характер ближнього оточення. У рідині встановлюється специфічний ближній порядок, поняття «міжплощинна відстань» втрачає сенс. Отже, і методи вивчення структури, засновані на рівнянні Вульфа — Брегга, до рідин непридатні.
Кількісний опис структури рідин і аморфних тіл здійснюється за допомогою радіальних функцій міжатомних відстаней, функцій атомної і молекулярної густини.
Нехай система з N однакових атомів займає об'єм V.
Виберемо в ньому два елементи об'єму dV1
і dV2
що фіксуються векторами R
1
і R
2
проведеними з деякої точки (мал. 1.2).

Якщо взаємне розташування атомів хаотичне, то вірогідність того, що атом 1 знаходиться в елементі об'єму dV1
а атом 2в той же час в dV2
, через незалежність їх положень рівна:
 (7) (7)
Хаотичне розташування атомів можливе лише в розріджених газах, коли власний об'єм атомів і сили взаємодії між ними можна не брати до уваги. У рідинах атоми не можуть знаходитися на довільній відстані один від одного, оскільки їх упаковка достатньо щільна. Вірогідність знаходження деякого атома в якій-небудь точці об'єму V залежить від того, в якій точці знаходиться інший атом. Такий вірогідний зв'язок між взаємним розташуванням атомів (їх кореляція) кількісно описується функцією W(R1
,R2
) Формулу (7) слід представити у вигляді:
 (8) (8)
Оскільки рідини ізотропні, то функція W залежить тільки від взаємної відстані між парою даних атомів:
W
(R
1
,R
2
) = W(|R
1
—R
2
|) = W(|R
12
|) = W(R) (1.9)
Рівність (8) можна виразити в іншій формі. Для цього сумістимо початок координат з центром атома 1 і опишемо навколо нього дві концентричні сфери радіусу R і R+dR .
Вірогідністьвиявлення атома 2 в сферичному об'ємі 4πR2
dRна відстані від R до R+dR від центру атома 1 рівна:
 (10) (10)
Функція W(R) задовольняє умові нормування:
 (11) (11)
показуючому, що сума вірогідності знаходження даного атома на всіх можливих відстанях від фіксованого рівна одиниці. Функція W(R) називається радіальною функцією розподілу атомів або молекул. Її значення визначають вірогідність виявлення якого-небудь атома на відстані Rвід фіксованого атома. Оскільки сили відштовхування перешкоджають взаємному проникненню атомів, то в інтервалі 0 ≤ R < 2r( r — радіус атома) функція W(R)= 0. При R→∞ вона прагне до одиниці.
Припустимо, що в шарі 4πR2
dRзнаходиться dn атомів. Тоді число атомів в одиниці об'єму цього шару визначиться формулою:
 (12) (12)
У різних сферичних шарах число атомів неоднакове, тому ρ — це функція відстані R. Кількість атомів в одиниці об'єму може змінюватися і унаслідок міграції атомів між сусідніми положеннями рівноваги, тому dn є середнім за час спостереження значенням. Отже,
dn = ρ(R)4πR2
dR(13)
Інтеграл  рівний числу атомів в об'ємі V, за винятком фіксованого атома. Якщо це число постійно, то в якості об'єму інтеграції приймається сфера, обмежена уявною поверхнею радіусу R0
в необмеженому однорідному просторі. рівний числу атомів в об'ємі V, за винятком фіксованого атома. Якщо це число постійно, то в якості об'єму інтеграції приймається сфера, обмежена уявною поверхнею радіусу R0
в необмеженому однорідному просторі.
 (14) (14)
Якщо ж N флуктує навколо середнього значення <N>, то
 (15) (15)
Її об'єм достатньо великий, щоб містити велике число атомів. При цьому передбачається, що атоми, розташовані поблизу поверхні сфери, мають те ж оточення, що і атоми, що знаходяться в її центрі. Співвідношення (14) і (15) визначають умови нормування функції атомної густини ρ(R) перша умова є точною для кристала, друга — для рідини. Зіставляючи функції (11) і (14), знаходимо
 (16) (16)
де ρат
— середнє число атомів в одиниці об'єму. Згідно цьому співвідношенню значення W(R) рівні відношенню істинної кількості атомів в одиниці об'єму до середньої атомної густини. Тому W(R) має сенс відносної радіальній функції розподілу. Функція W(R) є найважливішою і основною характеристикою структури атомарних рідин і аморфних тіл.
Гази.
Припустимо, що атоми — непроникні кульки. Тоді можна стверджувати, що вірогідність зближення двох атомів на відстань R<2r рівна нулю (мал. 1.3,а). Якщо густина газу дуже мала, то за межами сфери радіусу R = 2r розташування атомів по відношенню до фіксованого буде рівноімовірним (хаотичним). Число атомів в одиниці об'єму на цій відстані рівне середньому значенню ρат
а функція W(R)= 1. Якщо ж газ достатньо щільний, то при R = 2r функція W(R) має максимум, при R<2r вона прагне до нуля, а при R>2r— до одиниці (мал. 1.3,6).

Кристали.
У ідеальному кристалі за відсутності теплового руху атоми розташовані на фіксованих відстанях один від одного. Так, в кубічній гранецентрированій гратці 12 атомів знаходяться на відстані 2r, 6 — на відстані 2r(2)1/2
, 24 — на відстані 2r(3)1/2
12 — на відстані 4r і т.д. Вірогідність знаходження атома в проміжках між вказаними відстанями рівна нулю. Якщо уявити собі, що такий кристал обертається навколо одного з атомів, то центри інших атомів розташовуватимуться на сферах цих радіусів і жоден з них не виявиться між сферами. Радіальна функція W(R) має дискретний характер. Якщо зберегти за нею умову нормування (11), то розподіл атомів на відповідних сферах можна представити у вигляді:
 (17) (17)
де δ(R—Rk
)дельта-функція Дірака, числове значення якої визначають умовою0 при R ≠ Rk
δ(R—Rk)= {  (18) (18)
∞ при R = Rk
Тепловий рух змінює розміщення атомів в кристалі, їх центри декілька відхиляються від середнього положення рівноваги. Тому вертикальні лінії (мал. 1.3, в) слід замінити піками гаусової форми з напівшириною ∆R = (kT/β)1/2
де β — коефіцієнт квазіпружної сили.
Рідини. Амплітуда теплових коливань атомів біля положень рівноваги в рідині набагато більше, ніж в кристалах. До того ж атоми рідини, беручи участь в тепловому русі, безперервно обмінюються своїми найближчими сусідами. Якщо в думках прослідити за рухом якого-небудь атома в рідині, то можна переконатися, що за час спостереження він знаходитиметься на самих різних відстанях від фіксованого атома. Внаслідок цього функція W(R) буде безперервною. У інтервалі 2r<R<R0
вона осцилює щодо одиниці з амплітудою, що постійно зменшується. Максимуми цих осциляції відповідають вірогіднішим міжатомним відстаням, мінімуми — відстаням, на яких атоми знаходяться порівняно рідко. Послідовність максимумів W(R) відповідає послідовності рівноважних міжатомних відстаней в рідині і тому визначає ту впорядкованість на близьких відстанях, яка характеризує розташування атомів в речовині. При збільшенні відстані розташування атомів по відношенню до фіксованого рівноімовірне, причому функція W(R)→1, коли R>>r.
Функція W(R) є типовою для рідин з щільною упаковкою атомів (мал. 1.3,г). Якщо розташування атомів характеризується менш щільною упаковкою (вісмут, германій, сурма), то відповідна функція розподілу зображається кривою декілька іншого вигляду.
Зіставлення функцій W(R) для рідини і кристала показує, що у разі кристала максимуми цієї функції розділені проміжками, де, W(R)= 0, тоді як в рідині навіть перший пік не дозволений. Нерозв'язність піків радіальної функції зв'язана, очевидно, з розкидом рівноважних положень атомів і їх рухом трансляції. Якщо функція W(R) відома, то тим самим відомий і характер взаємного розташування частинок. Тому основною характеристикою молекулярної структури рідин є радіальна функція розподілу. Знаходження цієї функції для тієї або іншої рідини є найважливішою задачею структурного аналізу. Кількісними параметрами структури є координаційні числа, рівноважні міжатомні відстані, середні квадратичні зсуви атомів, а також відстань, на якому зникає кореляція в розташуванні частинок. Характеристиками структури рідин є також флуктуації концентрацій, густини і орієнтації молекул.
Дж. Кірквуд знайшов явний вид функції W(R) теоретично, виходячи із загальних принципів статистичної механіки. Імітуючи атоми твердими взаємодіючими кульками, він одержав формулу:
 (19) (19)
де A, α, β, і δ— постійні параметри. Наприклад, для аргону А = 9,51 Å; α= 0,30 Å -1
; σ =2,12 Å -1
; δ = —2,25. Функція W(R) обчислена по цій формулі, володіє такою ж залежністю від R як і експериментально визначувані функції розподілу для одноатомних щільно упакованих рідин.
Особливості розсіювання рентгенівського випромінювання, електронів і нейтронів
Рентгенографічні, електронографічні і нейтронографічні дослідження атомної і молекулярної структур рідин і аморфних тіл ґрунтуються на аналізі кутового розподілу інтенсивності розсіяного рентгенівського випромінювання, електронів і нейтронів. Розсіювання речовиною цих трьох видів випромінювань не однакове, що пояснюється відмінністю їх фізичної природи. Рентгенівське випромінювання розсівається електронами атомів і молекул. Процес розсіювання не характерний звичному віддзеркаленню або заломленню. Рентгенівське випромінювання, взаємодіючи з електронами, приводить їх в коливальний рух. Коливаючись з тією ж частотою, що і електричний вектор первинної електромагнітної хвилі, електрони породжують вторинне електромагнітне випромінювання, що розповсюджується на всіх напрямках. Інтенсивність розсіяного випромінювання, що фіксується в деякій точці, пропорційна електронній густині атомів і молекул.
Пучок прискорених електронів, що направляється на досліджувану речовину, розсівається електричним полем ядер і електронних оболонок атомів. Інтенсивність розсіювання електронів пропорційна електростатичному потенціалу атомів.
Нейтрони розсіваються ядрами атомів. При цьому пружне розсіювання повільних нейтронів використовується для вивчення атомної будови речовини, а непружне — для вивчення динаміки атомів і молекул.
Відмінність у взаємодії рентгенівського випромінювання електронів і нейтронів з речовиною враховується при розрахунку атомних амплітуд розсіювання, що є основними характеристиками розсіюючої здатності речовини. При розгляді ж розсіювання сукупністю зв'язаних атомів, іонів або молекул речовини механізм розсіювання не зачіпається. Беруть до уваги лише довжину хвилі використовуваного випромінювання, просторову конфігурацію частинок і відстань між ними.
Розсіювання рентгенівського випромінювання вільним електроном
Припустимо, що на вільний електрон направлений пучок паралельних монохроматичних рентгенівських променів, інтенсивність яких I0
.Електрон під дією вектора електромагнітної хвилі здійснює коливання, випромінюючи вторинні хвилі. Кутовий розподіл інтенсивності цих хвиль залежить від стану поляризації первинного рентгенівського випромінювання. Якщо вони поляризовані, то інтенсивність розсіювання одним електроном, що фіксується в точці на відстані L від електрона, виражається формулою
 (20) (20)
де m — маса електрона; e — його заряд; c— швидкість світла; φ— кут між напрямами коливання електрона і розсіюванням; e2
/(mc2
)— класичний радіус електрона, рівний 2,8•10-15
м; [e2
/(mc2
)]2—
його поперечник розсіювання. З (20) видно, що із збільшенням кута φ інтенсивність розсіювання збільшується, досягаючи найбільшого значення при φ = 90° (мал. 2.1).
Якщо первинний пучок рентгенівського випромінювання неполяризований, то формула для інтенсивності розсіювання одним електроном набуває вигляду
 (21) (21)
де (1+cos2
2θ)/2 = ρ(θ) — кутова залежність інтенсивності розсіяного рентгенівського випромінювання одним електроном названа поляризаційним чинником. Наявність цього множника у формулі (21) указує на те, що рентгенівське випромінювання при розсіянні частково поляризується. При гранично малих кутах розсіювання поляризаційний чинник рівний одиниці. Із збільшенням кута розсіювання цей чинник зменшується і при 2θ = 90° досягає значення, рівного 1/2. Таким чином, в різних напрямах інтенсивність розсіювання рентгенівського випромінювання одним електроном неоднакова. У напрямі первинного пучка і у зворотному напрямі інтенсивність розсіювання максимальна, а в перпендикулярному напрямі — мінімальна. Якщо у формулу (21) підставити значення постійних m, e і cто одержимо
 (22) (22)
Отже, один вільний електрон розсіює в одиницю тілесного кута 10-26
частину інтенсивності первинного пучка. Це значення розсіюваної інтенсивності приймають за умовну одиницю і використовують для нормування кривих інтенсивності. Приведені формули справедливі для будь-якого електромагнітного випромінювання, у тому числі і для видимого світла, довжини хвиль якого набагато більше розміри атомів і молекул.
Проте у разі видимого світла хвилі, випромінювані атомами і молекулами, не інтерферують одна з одною, оскільки під дією світлової хвилі всі електрони атомів здійснюють коливання в однаковій фазі. Атоми і молекули в полі світлової хвилі поводяться подібно елементарним електричним диполям. Картина істотно змінюється, якщо довжина хвилі падаючого випромінювання менше розміру атома. Тепер уже електрони в різних частинах атома коливаються в неоднакових фазах. Випромінювані ними хвилі приходять в точку спостереження з деякою різницею фаз і інтерферують одна з одною. Результат цієї інтерференції залежить від числа електронів в атомі і їх просторового розподілу. Досвід показує, що не все розсіяне випромінювання має ту ж довжину хвилі, що і первинне. Деяка його частина розсівається атомами некогерентно і участі в інтерференції не бере. У структурному аналізі використовується тільки когерентне розсіювання.
Розсіювання рентгенівського випромінювання вільним атомом
Електрони в атомі не можна розглядати як вільні, тому інтенсивність розсіювання рентгенівського випромінювання атомом не може бути одержана простим складанням інтенсивностейрозсіювання окремими електронами. При розрахунку інтенсивності розсіювання атомом необхідно враховувати різницю фаз вторинних хвиль, випромінюваних електронами в різних точках атома.
Розглянемо спочатку два електрони, що знаходяться в атомі в точках А і B на відстані r один від одного. Позначимо nіn0
— одиничні вектори у напрямі нормалі до фронту падаючої і розсіяної хвилі (мал. 2.2). Різниця ходу проміння, розсіяного електронами,
∆l = |AD|—|CB| = rcosβ—rcosβ0
= rn
—rn
0
= r
(n
—n
0
) (23)
де β0
—кут між напрямом падаючого променя і вектором r
; β—те ж, для розсіяного променя. Якщо r
(n
—n
0
) = 2k(λ/2) те розсіяні хвилі підсилять одна одну, якщо ж r
(n
—n
0
) = (2k + 1) λ/2 те ослаблять. Рівняння (23) виражає умову інтерференції розсіяних хвиль
r(cosβ—cosβ0
) = kλ(24)

При k = 1 і β0
= 90° одержуємо cos β =λ/r . Оскільки cosβ ≤ l, то звідси витікає, що інтерференція розсіяних хвиль виникає лише у разі, коли довжина хвилі менше відстані між частинками, що розсіюються. Якщо ж λ > r, інтерференція розсіяних хвиль не відбувається, Помноживши (23) на хвильове число 2π/ λ, одержимо вираз для різниці фаз хвиль, розсіяних двома електронами атома,
∆φ = (2π/ λ)r
(n
—n
0
) (25)
Припустимо, що електрони в атомі розподілені безперервно. Виділимо в ньому елемент об'єму dVi
і позначимо ρe
(ri
) — електронну густину в точці на відстані ri
від центру атома. Тоді число електронів, що знаходяться в об'ємі dVi
визначиться величиною ρe
(ri
) dVi
а амплітуда хвилі, розсіяної ними, — добутком

Сумарна амплітуда хвиль, розсіяних атомом,
 (26) (26)
Вектор n
—n
0
співпадає з напрямом нормалі до площини, що відображає рентгенівське випромінювання. У атомі відбиваючих площин зрозуміло ні. Проте поняттям «нормаль до відбиваючої площини», ми користуватимемося і в даному випадку, оскільки вектор n
—n
0
визначає напрям осі, від якої відлічується полярний кут.
Якщо кут між напрямами первинного пучка і уявною площиною, що відображає, позначити θ, то кут розсіювання 2θ.
Очевидно, що |n
—n
0
| = 2sinθ(мал.2.2) Позначаючи α — кут між векторами r
і n
—n
0
, одержимо для різниці фаз розсіяних хвиль вираз
 (27) (27)
де S = (4π/λ)sinθ Параметр S, залежний від довжини хвилі і кута розсіювання, зустрічається в структурному аналізі і в теорії твердого тіла. Він пов'язаний з міжплощинною відстанню d для площин кристалічних граток, від яких походить віддзеркалення першого порядку під кутом θпри довжині хвилі λ.Згідно умові віддзеркалення 2dsinθ= λ , маємо
2sinθ/ λ = 1/d або 4πsinθ/ λ = 2π/d, тобто S = 2π/d(28)
З другого боку, параметр S пов'язаний з хвильовим вектором розсіяної хвилі співвідношенням
S = 2|k
|sinθ(29)
а також з вектором оберненої гратки рівністю
S = 2π|r
*
| (30)
Підставляючи (27) в (26), одержимо для амплітуди розсіювання атомом вираз
 (31) (31)
Щоб додати йому конкретніший вигляд, припустимо, що розподіл електронів в атомі сферично симетричний і ρ(r) залежить тільки від модуля вектора r
, але не від його напряму. В цьому випадку елемент об'єму dV = r2
drsinαdαdφ. Вираз (31) можна написати у вигляді
 (32) (32)
Інтегруючи (32) по α і φ, одержимо
 (33) (33)
де 4πr2
ρe
(r)dr—
число електронів в сферичному шарі атома між радіусами r і r + dr .
Функція
 (34) (34)
характеризує розсіюючу здатність атома і називається атомною амплітудою, а F2
(S) — атомним чинником розсіювання. Числове значення F(S) показує, в скільки разів амплітуда розсіювання атомом в даному напрямі більше амплітуди розсіювання одним електроном. При S → 0 функція sinSr/(Sr) → 1; значення F(S) при нульовому куті розсіювання рівне числу електронів атома:
 (35) (35)
Отже, чим вище порядковий номер хімічного елементу, тим більше числове значення F(S). Із збільшенням параметра S функція F(S) монотонно убуває.
Щоб обчислити атомну амплітуду F(S) теоретично, потрібно знати просторовий розподіл електронної густини в атомі. Згідно квантової теорії, вірогідність знаходження електрона в точці на відстані r від центру атома визначається хвильовою функцією |Ψ|2
.
У разі атома водню
 (36) (36)
де r1
— радіус першої боровськой орбіти атома Н. Відповідний вираз для F(S) приймає вигляд
 (37) (37)
звідки
 (38) (38)
Ця формула показує, що атомна амплітуда розсіювання залежить тільки від S =2ksinθ. Як ρ(r) функція F(S) сферично симетрична. Відмінність між F(S) і ρ(r)полягає у тому, що функція ρ(r)описує розподіл електронної густини в звичному просторі, F(S) представляє цей розподіл в k— просторі, тобто просторі хвильових векторів. Числові значення F(S) для атомів деяких елементів приведені в довідкових таблицях. Знаючи F(S) можна написати вираз для інтенсивності розсіювання атомів:
 (39) (39)

Найбільший внесок в когерентне розсіювання вносять внутрішні електрони атома. Зовнішні електрони атома обумовлюють інтенсивне когерентне розсіювання при малих кутах. Це виразно видно з мал. 2.3,а, на якому представлено радіальний розподіл електронної густини ls2
2s2
2p6
3s2
3p6
електронів іона К+
. Там же показане (мал. 2.3,6) відповідне їм f-криві розсіювання. З малюнка видно, що чим далі від ядра знаходиться дана група електронів, тим швидше убуває відповідна їй f-функція з кутом розсіювання. Дійсно, порівнюючи f-криві для ls2
-,2s2
- і 3s2
электронов іона К+
, бачимо, що значення f1
s
, обумовлене розсіюванням ls2
-электронов (r1
= 0,03 Å), майже не змінюється з кутом розсіювання; f2
s
— крива, обумовлена розсіюванням 2s2
-электронами (r2
= 0,18 Å), монотонно спадає, тоді як для f3
s
-кривої (r3
= 0,6 Å) характерне швидке убування з переходом в область негативних значень з подальшою сильно затухаючою осциляцією біля осі абсцис. Амплітуда сумарного розсіювання іона К+
F(S) = f1s
(S) + f2s
(S) + f2p
(S) + f3s
(S) + f3p
(S) (40)
Відзначимо, що

є інтегралом Фурье, який має властивість оборотності.

Це дозволяє визначити функцію радіального розподілу електронної густини атома за даними про амплітуду розсіювання на цьому атомі, тобто перейти від зворотного простору до звичного координатного простору. При цьому
 (41) (41)
На мал. 2.4 представлена залежність електронної густини від відстані в атомі неону. Як видно, дані експерименту цілком відповідають теоретичним розрахункам.
В розсіяному випромінюванні присутні хвилі із зміненою довжиною хвилі. Вони виникають в результаті ефекту Комптона, тобто зіткнень первинних фотонів рентгенівського випромінювання із зовнішніми слабкозв'язанними електронами атомів. Фотон при зіткненні з електроном віддає йому частину енергії і імпульсу, і передає кінетичну енергію mυ2
/2(мал. 2.5). Відхиляючий від первинного напряму, фотон має вже меншу енергію і менший імпульс і має велику довжину хвилі. Нехтуючи релятивістськими ефектами, запишемо:
а) рівняння збереження енергії
 (42) (42)
б) рівняння збереження імпульсу
 (43) (43)
Тоді зміна довжини хвилі фотона при некогерентному розсіянні

З цієї формули видно, що у міру збільшення кута розсіювання значення ∆λ, зростає від нуля до 0,048 Å.
У структурному аналізі має істотне значення не стільки зміна довжини хвилі при розсіянні рентгенівського випромінювання, скільки внесок некогерентного розсіювання в сумарну інтенсивність розсіювання досліджуваної речовини.

Некогерентне розсіювання дає безперервний фон, інтенсивність якого зростає з кутом розсіювання. При великих значеннях S некогерентне розсіювання від елементів з малим атомним номером може перевершувати когерентне у декілька разів (мал. 2.6). Тому воно завжди віднімається із загального розсіювання. Звичний спосіб обліку поправки на некогерентне розсіювання полягає в обчисленні його величин теоретично по одній з формул, виведених для цієї мети. Одна з них має вигляд
 , ,
 , ,
де Z
— атомний номер елементу.
Таким чином, повна інтенсивність незалежного розсіювання одним
атомом складається з когерентного і некогерентного доданків:
I(S)= IK(S)+ IHK(S) (44)
Розсіювання електронів вільним атомом
Застосування методу дифракції електронів для дослідження молекулярної структури речовини засноване на хвильових властивостях цих частинок. Пучок електронів, що розповсюджується у напрямі осі X, можна представити плоскою монохроматичною хвилею, описуваною хвильовою функцією

Взаємодіючи з електричним полем атома, ця хвиля частково розсівається. На відстані L від центру атома розсіяна хвиля представляється у вигляді
 (45) (45)
де fe(S) — атомна амплітуда розсіювання електронів, що має розмірність довжини.
Результуюча електронна хвиля представляється суперпозицією падаючої плоскої і розсіяної сферичної хвиль:
 (46а) (46а)
або
Ψ = Ψ0
+ η (46б)
де А і B — деякі постійні.
Тут η << Ψ0
, оскільки розсіювання складає лише незначну частку первинного пучка. Результуюча хвильова функція Ψ електрона, рухаючегося в електричному полі атома, знаходиться з рівняння Шредінгера:
 (47) (47)
де m — маса розсіюючого електрона, E — його повна і U(r) —-потенційна енергія в електричному полі атома, h — постійна Планка. Передбачається, що U(r) швидко убуває із зростанням відстані r від ядра. Підставимо (46б) в (47). Враховуючи, що хвильова функція Ψ0
плоскої хвилі задовольняє рівнянню руху електрона поза атомом (∆Ψ0
+ k2
Ψ0
= 0), і нехтуючи добутком U(r)η як величиною другого порядку малості, одержимо
 (48) (48)
деk2
= 8π2
mE/h2
= (2π/λ)2
Вираз (48) аналогічно рівнянню Пуассона. Його рішення має вигляд
 (49) (49)
де r — відстань від центру атома до електрона; L — відстань від центру Oатома до точки спостереження A;l = L—rn
; (r
n
) — проекція вектора r
на напрям розсіювання n
(мал. 2.7). Якщо точка спостереження знаходиться на великій відстані від центру атома, то в знаменнику (49) можна замінити l на L.
Тоді
(50) 
Зіставляючи це рівняння з (45), можна записати вираз для атомної амплітуди розсіювання електронів:
 
 (51) (51)
Переходячи до сферичних координат і інтегруючи (51) по α і φ, одержуємо
 (52) (52)
Цей вираз нагадує атомний чинник для рентгенівського випромінювання
 (51) (51)
Зіставляючи вирази (52) і (51), помічаємо, що амплітуда розсіяної електронної хвилі пропорційна потенційній енергії електрона в полі атома, тоді як амплітуда розсіювання рентгенівського випромінювання пропорційна електронній густині атома.
Для безпосередніх обчислень атомних амплітуд розсіювання електронів зручно у формулі (51) виразити потенційну енергію U(r) через електронну густину ρe
(r). Запишемо в явному вигляді вираз для U(r).
Електростатичний потенціал в будь-якій точці атома складається з потенціалу позитивно зарядженого ядра і потенціалу електронної оболонки. Потенціал в точці r, створюваний зарядом ядра, рівний —
Ze/r.
Щоб визначити потенціал, створюваний в тій же точці зарядом електронної оболонки атома, розглянемо елемент об'єму dV1
на відстані r1
від центру О атома (мал. 2.8). Заряд, зосереджений в цьому об'ємі, рівний eρe
(r1
)dV1
.
Потенціал в точці r, створюваний цим зарядом, eρe
(r1
)dV1
/|r
—r
1
|. Потенціал в тій же точці, створюваний всією електронною оболонкою атома, представиться як

Загальна потенційна енергія електрона усередині атома виразиться формулою
 (52) (52)
Підставляючи цей вираз в рівняння (52), одержимо

(53)
Візьмемо перший інтеграл
 (54) (54)
якщо, як і раніше, покласти dV = r2
drsinαdαdφ. Щоб обчислити другий доданок, замінимий в ньому порядок інтеграції:

Інтеграл

якщо eiSrcosα
= ρе
(r), тобто він може розглядатися як потенціал, створюваний в точці r електричним зарядом, розподіленим в просторі з густиною ρе
(r), З другого боку, потенціал φ(r) пов'язаний з густиною ρе
(r), рівнянням Пуассона
Δφ(r) = — 4π ρе
(r) = — 4πei S r
(55)
Інтегруючи (55) по r, одержимо
φ(r) = 4πei S r
/S2
(56)
Отже, (57) (57)
Другий доданок в (53) можна представити у вигляді

Інтегруючи по α і φ і опускаючи індекс при r, одержимо

Атомна амплітуда розсіювання електронів визначається формулою
 (58) (58)
Або (59) (59)
Використовуючи значення атомних амплітуд розсіювання рентгенівського випромінювання, можна по цій формулі обчислити fe(S) для будь-якого елементу. Звівши (59) в квадрат, одержимо інтенсивність когерентного розсіювання окремим атомом:
 (60) (60)
Підставляючи числові значення постійних, знайдемо

Когерентне розсіювання електронів складається з ядерного і електронного: член, що містить r2
, визначає частку інтенсивності розсіювання ядром, член з F2
(S) — інтенсивність розсіювання оболонкою атома, нарешті, член, що містить ZF2
(S) визначає інтенсивність розсіювання електронною оболонкою і ядром. Загальна інтенсивність розсіювання електронів убуває обернено пропорційно до S4
. У разі рентгенівського випромінювання інтенсивність розсіювання спадає обернено пропорційно до S. Зменшення інтенсивності з кутом розсіювання пояснюється тим, що довжина хвилі цих випромінювань менше розмірів атомів. Внаслідок цього відбувається інтерференція хвиль, розсіяних кожним атомом окремо.
Порівняння (60) з (39) показує, що розсіювання електронів тими ж атомами майже в 106
разів більше розсіювання рентгенівського випромінювання. Цим обумовлюється швидкість отримання електронограм. Експозиції електронографічних досліджень вимірюють секундами, тоді як при рентгенографічних — хвилинами і годинами. До того ж для спостереження картини дифракції електронів достатньо узяти плівку в 200—300 Å, тоді як товщина шаруючи речовини при рентгенографічних дослідженнях близько 1 мм.
При розсіянні електронів разом з когерентними, розповсюджуються електрони, що втратили частину своєї енергії унаслідок непружного розсіювання на атомах. Це розсіювання викликає фон, інтенсивність якого обчислюють по формулі
 (61) (61)
де IHK
(S) — інтенсивність некогерентного розсіювання рентгенівського випромінювання.
Розсіювання повільних нейтронів на вільному ядрі
Застосування нейтронів для дослідження атомномолекулярної структури речовини засноване на явищі дифракції (розсіювання) цих частинок. Використовують повільні нейтрони з энергией 2 •10-
1
— 2•10-
3
еВ, що згідно формулі
λ = h/(2mE)1/2
(62)
відповідає довжині хвилі 0,5 — 6,0 Å.
Через відсутність у нейтронів електричного заряду їх розсіювання інше, ніж у рентгенівського випромінювання і електронів. Процес розсіювання нейтронів не залежить від заряду ядер, а визначається їх складом і спином.
Розсіювання нейтронів пояснюється взаємодією їх з ядрами. Воно характеризується ефективним перерізом розсіювання, визначуваним як відношення числа нейтронів, що відхилюють одним ядром за одиницю часу, до числа нейтронів, падаючих за той же час на одиницю площі шаруючи речовини: σ =Δn/n З цього визначення виходить, що σ має розмірність площі. Дійсно, оскільки [Δn] = 1/T, [n] =1/(TL2
) то [σ] = L2
. Перетин розсіювання нейтронів можна виразити через хвильову функцію падаючих і розсіяних хвиль. Якщо  — хвильова функція падаючої на ядро плоскої нейтронної хвилі, а — хвильова функція падаючої на ядро плоскої нейтронної хвилі, а  — хвильова функція сферичної розсіяної хвилі, то згідно сказаному повний переріз розсіювання ядром — хвильова функція сферичної розсіяної хвилі, то згідно сказаному повний переріз розсіювання ядром
 (63) (63)
де fn
— амплітуда когерентного розсіювання нейтронів. Оскільки fn
має розмірність довжини, то її називають також довжиною розсіювання. Відмітною особливістю розсіювання повільних нейтронів є ізотропна по всіх напрямах, незалежність його перетину від енергії налітаючих нейтронів. Це пояснюється тим, що довжина хвилі повільних нейтронів (λ≈10-10 м
) велика в порівнянні з радіусом дії силового поля ядра (r ≈10-15 м
), а їх енергія мала в порівнянні з енергією зв'язку усередині ядра.
Для детальнішої характеристики взаємодії нейтронів з ядром вводять поняття диференціального переріза розсіювання dσ,
визначуваного як кількість нейтронів, розсіяних усередині тілесного кута dΩ. Диференціальний переріз залежить від кута розсіювання. Дійсно, якщо на ядро, що покоїться, направити пучок нейтронів, то залежно від того, на якій прицільній відстані від ядра вони пролітають, кут їх розсіювання буде неоднаковий. Деякі налітаючі нейтрони розсіваються під кутом, близьким до 180°, інші — під дуже малими кутами.
Отримання і інтерпретація даних по розсіянню нейтронів з метою визначення структури речовини засновані на вимірюванні диференціального переріза розсіювання залежно від кута θі енергії En
налітаючих нейтронів.
Дослідження показують, що взаємодія нейтрона з речовиною може привести не тільки до розсіювання, але і до захоплення його ядром і утворенню проміжного ядра з подальшим випуском нейтрона. Який з цих процесів переважає, залежить від енергії падаючого нейтрона і властивостей ядра.
Отже, в загальному випадку ядерне розсіювання повільних нейтронів є накладенням потенційного і резонансного розсіювання. Загальна амплітуда розсіювання без урахування спину ядра представляється у вигляді двох доданків:
  (64) (64)

де En
— енергія падаючого нейтрона; Ep
— енергія, якою повинен володіти нейтрон, щоб викликати резонанс в складеному ядрі; i—число ізотопів; Гn
— нейтронна ширина енергетичного рівня, пов'язана з вірогідністю розсіювання нейтрона, падаючого на ядро- мішень; Г — ширина резонансного максимуму на половині його висоти, рівної σт
(мал. 2.9) (тут σр
— перетин при резонансі, тобто при E = Ep
).
Диференціальний переріз розсіювання на вільному ядрі визначається по формулі
 (65) (65)
За відсутності у ядра резонансних рівнів, достатньо близьких до енергії падаючого нейтрона, резонансним членом можна знехтувати. В цьому випадку амплітуда розсіювання визначатиметься чисто потенційним членом, який завжди позитивний і рівний радіусу r ядра:
dσi
= f2
n
dΩσi
= 4πr2
(66)
На підставі останньої формули можна укласти, що потенційне пружне розсіювання повільних нейтронів відбувається як би на непроникних сферах того ж радіусу, що і ядро. Оскільки радіус ядра r = 1,5•10-15
(A)1/3
[м] [м], де A—атомна маса ядра, те значення σi
може бути обчислене для будь-якого елементу. Співвідношення (66) добре виконується для важких елементів. Для легких атомів спостерігається відхилення від цієї залежності.
При наближенні енергії падаючих нейтронів до значення енергії резонансного рівня ядра другий доданок в (64) стає достатньо великим, щоб переважати над потенційним членом. При цьому різниця E—Ep
може бути як позитивною, так і негативною. Для H, Li і Мn резонансний член, будучи негативним, переважає над потенційним, приводячи, таким чином, до негативної амплітуди розсіювання. Якщо ядро володіє спином j, то результат складання його із спином падаючого нейтрона, рівним ±1/2, може привести до утворення складених ядер із спинами відповідно j + 1/2 і j — 1/2; В цьому випадку розсіювання повільних нейтронів на вільному ядрі описуватиметься не одній, а двома амплітудами розсіювання: f+
іf—
.Перша амплітуда відповідає паралельній взаємній орієнтації спинів ядра і падаючого нейтрона, друга — антипаралельної орієнтації спинів. При цьому диференціальний переріз розсіювання
 (67) (67)
Множники при f2
+
і f2
—
. визначають вірогідність реалізації різних станів спинів системи з нейтрона і ядра при їх зіткненні.
Формула (67) показує, що розсіювання повільних нейтронів на вільних ядрах повністю визначається значенням амплітуд f+
і f —
. які можуть бути знайдені експериментально. Якщо в розсіянні нейтронів бере участь система зв'язаних ядер, то амплітуда розсіювання на вільному ядрі повинна бути замінена амплітудою розсіювання на зв'язаному ядрі. У разі одноатомної речовини fn
→ b = f(1 + 1/A). Дослідження показують, що амплітуди розсіювання повільних нейтронів для різних ядер знаходяться в інтервалі від 0,3•10-14
до 1•10-14 см, що відповідає інтегральному перетину розсіювання
σ ≈ 10-28
м2
. Це майже на два порядки більше відповідної величини для рентгенівського проміння.
Якщо в розсіянні бере участь не одне ядро, а деякий колектив ядер, то розсіювання повільних нейтронів матиме когерентну і некогерентну складові. Когерентне розсіювання викликається впорядкованим розташуванням ядер. У некогерентному розсіянні ядра беруть участь неузгоджено, що говорить про безлад в розташуванні ядер. Наявність у нейтрона магнітного моменту приводить до магнітного розсіювання нейтронів речовиною. Якщо магнітні моменти атомів або іонів розсіювача орієнтовані хаотично (парамагнетіки), то магнітне розсіювання має дифузний характер. Якщо ж останні мають впорядковану орієнтацію (феромагнетики і антиферомагнетики), то магнітне розсіювання повільних нейтронів є когерентним і разом з ядерним когерентним розсіюванням вносить внесок в загальне розсіювання. Аналіз даних по розсіянню нейтронів дає пряму інформацію про розподіл і орієнтацію магнітних моментів атомів в досліджуваній речовині, що неможливо одержати інакше. На мал. 2.10 показані атомні амплітуди когерентного розсіювання рентгенівського випромінювання, електронів і нейтронів. Найсильніша залежність атомної амплітуди від кута розсіювання у електронів, менш сильна — у рентгенівського випромінювання і зовсім вона відсутня у повільних нейтронів. Це враховується при постановці і проведенні структурних досліджень.

Істотно, що амплітуди розсіювання рентгенівського випромінювання і електронів однакові для всіх ізотопів даного елементу, тоді як амплітуди розсіювання нейтронів fn
для різних ізотопів різні. Завдяки цьому повільні нейтрони служать виключно зручним засобом вивчення структури твердих тіл і рідин, що містять атоми з дуже близькими або достатньо далекими порядковими номерами; вони практично незамінні в структурних дослідженнях сполук, що містять, водень дозволяючи фіксувати положення атомів водню і довжину водневих зв'язків.
Відзначимо, що з трьох видів випромінювань, вживаних для дослідження структури рідин, найбільш підходить рентгенівське. Щоб в цьому переконатися, порівняємо енергію нейтрона і рентгенівського фотона, а також час прольоту ними відстані порядку міжатомного, тобто 10-10
м. При цьому
Eф
=hc/λEn
=h2
/(2mλ2
)
звідки
Eф
=2mcλ/h = 105 En
(68)
Оскільки швидкість фотона c ≈ 108
м/с, а швидкість нейтрона υn
=
(3kT/m)1/2
= 103
м/с, той час проходження ними відстані порядка 10-10 м
складає 10-18
с для фотона і 10-13
с для нейтрона. Отже, енергія рентгенівських фотонів майже в 105
разів більше, ніж енергія нейтронів при тій же довжині хвилі. У стільки ж разів менше тривалість взаємодії фотона з атомом. Тому для рентгенівського випромінювання непружне розсіювання атомів не виконує ролі, для нейтронів же воно складає значну частину загального розсіювання, що ускладнює методику дифракційного експерименту. Разом з цим слабке поглинання нейтронів дозволяє одержувати діфрактограми від рідких металів, сильно поглинаючих рентгенівське випромінювання. Застосування до рідин електронографічних досліджень зв'язане з рядом важкоусуваємих побічних ефектів. Електрони є зручним засобом вивчення будови молекул газів, структури кристалічних і аморфних тіл.
Розсіювання однаковими атомами
Розглянемо розсіювання рентгенівського випромінювання, електронів і нейтронів сукупністю атомів одного елементу (зріджені інертні гази, розплавлені метали, напівметали і діелектрики). Виведемо рівняння, що зв'язує кутовий розподіл інтенсивності розсіяного випромінювання з радіальною функцією розподілу W(R) якаописує ближній порядок в розташуванні атомів. Припустимо, що паралельний пучок монохроматичного проміння довжиною хвилі λ направлений на зразок досліджуваної речовини, миттєве положення атомів якого визначається векторами R
1
,R
2
,…R
N
щодо довільно вибраного початку відліку. Позначимо F1
,F2
,…FN
— атомні амплітуди розсіювання; N —
число атомів, що беруть участь в розсіянні. Сумарну амплітуду хвилі, розсіяної даною конфігурацією атомів, можна представити у вигляді
 (69) (69)
Відповідну інтенсивність визначимо множенням виразу (69) на його комплексно-зв'язану величину:

або
 (70) (70)
де Ie
інтенсивність, віднесена до інтенсивності розсіювання одним електроном; Rj
— Rk
— векторна міжатомна відстань. Подвійна сума містить N2
членів. Серед них є N членів, для яких j ≠ k. Для кожного такого члена експоненціальний множник звертається в одиницю. Інші N2
— Nчленів залежать від взаємного розташування атомів. Оскільки, по припущенню, всі атоми системи однакові, вираз (70) приймає вигляд
 (71) (71)
Воно визначає інтенсивність розсіяного випромінювання, обумовленого миттєвим розташуванням атомів. Проте дифракційний експеримент дає не миттєву, а середню за час експозиції картину розсіювання.
Для того, щоб теоретично знайдений кутовий розподіл інтенсивності і одержане з досвіду відповідали один одному, необхідно всі члени подвійної суми в (71) усереднити по всіх можливих положеннях атомів в опромінюваному об'ємі зразка. Результат усереднювання залежатиме від того, чи є міжатомний вектор R
jk
= R
j
— R
k
постійним по модулю або ж що безперервно змінюється від точки до точки. Випадок R
jk
= constвідноситься до молекули, другої — до речовини з безперервним розподілом атомів. Досліджуємо газ, молекули якого складаються з n атомів. Якщо тиск газу не дуже великий, то за кінцевий проміжок часу всі орієнтації молекул зустрічатимуться однаково часто. Отже, щоб одержати повну інтенсивність розсіювання в газі, потрібно визначити середнє значення інтенсивності для однієї молекули і помножити його на число молекул газу.
Щоб визначити середнє значення I(S), розглянемо в молекулі атоми j і k.Сумістимо початок координат з центром атома j. За вісь відліку кута α приймемо вектор n
— n
0
паралельний осі Z.
Тоді вірогідність того, що напрям вектора R
jk
складає з осями координат кути, укладені між α і α + dα, φ і φ + dφ, рівна відношенню елементу сферичної поверхні до поверхні сфери:
 (72) (72)
Умножаючи (71) на (72) і інтегруючи по кутах α і φ, знайдемо для однієї молекули формулу вперше одержану Дебаєм.
 (73) (73)
Вона описує зв'язок між кутовим розподілом інтенсивності розсіювання окремими молекулами і їх структурою. Якщо молекули газу двухатомні, то інтенсивність розсіювання ними рівна
 (74) (74)
При малих значеннях S інтенсивність I(S)наближається до 4F2
, а при великих S — до 2F2
. У області проміжних значень S крива має максимуми і мінімуми, положення яких визначимо, прирівнявши нулю похідну функції (74). Припускаючи, що атоми розсіюють як точки, що справедливе для нейтронів, одержимо рівняння tgSR = SR.
З його рішення виходить, що перший максимум I(S)з'являється при S1
R1
= 2,459π = 7,73звідки
R1
= 7,73/S1
(75)
Насправді атоми розсіюють рентгенівське випромінювання і електрони не як точки і функція F2
(S), що фігурує як співмножник у формулі (74), швидко убуває у міру зростання S. В результаті максимуми на кривій розсіювання стають менш чіткими, їх положення зміщується у бік великих S. Тому, щоб по формулі (75) обчислити відстань між атомами в двоатомній молекулі, необхідно розділити інтенсивність, заміряну для кожного кута, на атомний чинник, відповідний цьому куту. При цьому виходить функція інтенсивності а(S)= 1 + sinSR/(SR) перший максимум якої описується формулою (75). Якщо молекула містить більше двох атомів, то експериментальна крива інтенсивності визначиться сумою кривих, описуваних рівнянням (74). При цьому положення першого максимуму може не відповідати значенню R1
. Для рідин і аморфних тіл обчислення середнього значення подвійної суми у виразі (71) роблять за допомогою радіальної функції розподілу W(R),
пов'язаної з вірогідністю знаходження атома j в елементі об'єму dVj
а атома k— в елементі dVk
,
співвідношенням
 (76) (76)
де V — об'єм розсіюючої частини зразка; Rjk
— відстань між парою атомів.
Середнє значення часток інтенсивності, що вносяться парами атомів j і k,
виходить при множеннях кожного члена в подвійній сумі на (76) і інтеграціях по елементах об'єму як для dVj
так і для dVk
. Отже,
 (77) (77)
При збільшенні Rjk
функція W(Rjk
)→1, тому її зручно уявити у вигляді
W(Rjk
) = [W(Rjk
) — 1]+1 (78)
Припускаючи, що всі N(N — 1) членів подвійної суми рівні між собою, і нехтуючи одиницею в порівнянні з N,
маємо
 (79) (79)
Або<
I> =
NF2
(1 + NX1
+ NX2
) (80)
Розглянемо інтеграл
 (81) (81)
Інтеграція по Vj
розповсюджується на весь об'єм розсіюючої частини зразка, який можна прийняти за сферу радіусу L. Що ж до об'єму Vk
той його аналітичний вираз залежить від взаємного розташування атомів. Але оскільки функція W(Rjk
) сферично симетрична і при Rjk
> Rk
рівна одиниці, можна припустити, що Vk
має форму сфери, радіус Rk
якої визначає протяжність ближньої впорядкованості атомів.
Щоб обчислити подвійний інтеграл (81), припустимо, що вірогідність знаходження атома усередині об'єму V скрізь однакова. Тоді
 Сумісний центр атома j з початком координат. Положенняатома kпо відношенню до атома j визначатиметься відстанню R і кутами α і φ. Вираз (81) перетвориться до вигляду Сумісний центр атома j з початком координат. Положенняатома kпо відношенню до атома j визначатиметься відстанню R і кутами α і φ. Вираз (81) перетвориться до вигляду
 (82) (82)
Інтегруючи (81), одержимо
 (83) (83)
Подвійний інтеграл обчислюється точно в припущенні, що розсіююча частина зразка має форму сфери радіусу L.
 (84) (84)
Підінтегральний вираз розпадається на два множники, одні з яких залежить від координат атома j а інший — від координат атома k.При цьому кожна інтеграція розповсюджується на весь об'єм V.Маємо
 (85) (85)
Підставляючи в(80) формули(83) і(85) знаходимо що усереднена інтенсивність розсіювання рівна
 (86) (86)
Оскільки функція W(R)= 1 при R ≥ Rk
то межі інтеграції від 0 до ∞
можна замінити межами від 0 до Rk
. Враховуючи, що W(R) = Nρат
(R)/V, а N/V = <ρат
> одержимо
 (87) (87)
Перший доданок визначає інтенсивність розсіювання окремими атомами за відсутності інтерференції між ними; друге — розподіл інтенсивності розсіювання за наявності інтерференції, обумовленої ближнім порядком в розташуванні атомів. Третій доданок визначає інтенсивність розсіювання у області дуже малих кутів. Числове значення цього доданку залежить від розміру і форми зразка і не залежить від його внутрішньої структури. Дійсно, максимальне значення функції φ(SL)= 3(sinSL—SLcosSL)/(SL)3
дорівнює одиниці при SL = 0. Із зростанням SL функція φ(SL) здійснює сильно затухаючі осциляції щодо нульових значень, визначуваних рівнянням SL = tgSL, тобто при SL рівних 4,49; 7,74. При SL> 4,49 значення φ(SL) малі в порівнянні з одиницею. З рівності S= 4,49/L витікає, що для зразків порядка 0,1—0,2 см значення S = 4,5•10-7
Å -1
. Малокутове розсіювання на зразках таких розмірів співпадає з первинним пучком. Його інтенсивність не може бути заміряна за допомогою звичних засобів. Це розсіювання експериментально виявляється в тих випадках, коли в досліджуваній речовині є флуктуації, колоїдні частинки або макромолекули розміром до 103
Å. Таким чином, за винятком малокутового розсіювання, інтенсивність, вимірювана експериментально, визначається рівнянням
 (88) (88)
Щоб написати аналогічні рівняння для випадку розсіювання електронів тією ж речовиною, слідує атомну амплітуду розсіювання рентгенівського випромінювання замінити на атомну амплітуду розсіювання електронів, залишивши решта членів без змін. Якщо при дослідженні застосовуються нейтрони, то рівняння (88) можна представити у вигляді
 (89) (89)
де bК
— амплітуда когерентного розсіювання нейтронів зв'язаними ядрами, усереднена по станах спинів і ізотопах даного елементу. Застосовуючи до рівнянь (88) і (89) Фурье-перетворення, одержимо:
 (90) (90)
 (91) (91)
Ці рівняння лежать в основі вивчення структури атомарних рідин і аморфних тіл.
Параметри, визначувані по кривих інтенсивності
Безпосереднім результатом рентгено-, електроно- і нейтронографічних досліджень рідин і аморфних тіл є інтерференційна картина. У разі одноатомних рідин і аморфних тіл вона несе інформацію про ближній порядок в розташуванні атомів. Картина розсіювання молекулярними рідинами і аморфними тілами відображає атомний склад молекул, їх конфігурацію і взаємне розташування. Задача дослідження полягає в тому, щоб по інтерференційній картині відтворити просторову структуру речовини, встановити зв'язок між структурою і фізичними властивостями.
Для опису структури і структурно – чутливих властивостей рідин і аморфних тіл використовується не вся інтенсивність розсіювання, а лише її інтерференційна (структурна) частина
 (92) (92)
Числові значення структурного чинника а(S) рівного відношенню спостережуваної інтенсивності когерентного розсіювання до інтенсивності незалежного розсіювання того ж числа атомів. При великих S, а також в тих випадках, коли розподіл атомів хаотичний, функція а(S)= 1. Під час переходу речовини із стану з неврегульованим розташуванням атомів в стан з впорядкованим їх розташуванням відбувається перерозподіл інтенсивності, посилення її в одних напрямах і ослаблення в інших. Функція а(S) осцилює з амплітудою, що поступово зменшується, біля одиниці, залишаючись позитивною при всіх значеннях S(мал. 2.11).

Згідно (92) послідовність максимумів а(S) визначається послідовністю максимумів функції sinSR/(SR).
Ця функція має максимуми при значеннях SR, рівних 7,73; 14,06; 20,46; ... Отже,
R1
= 7,73/(S1
)max
= 14,06/(S2
)max
= 20,46/(S3
)max
= … (93)
Звідси видно, що у разі одноатомних рідин і аморфних тіл середня відстань від фіксованого атома до його найближчих сусідів визначається по значенню S, відповідному будь-якому максимуму інтерференційної функції а(S). Це означає, що визначаючим в утворенні картини розсіювання одноатомними рідинами і аморфними речовинами є найкоротша міжатомна відстань R1
що повторюється в різних порядках інтерференції.
Характерний, що значення R1
визначуване по кривій а(S), близько до значення істинної найкоротшої міжатомної відстані, тільки для рідин з щільною упаковкою атомів (зріджені, інертні гази; типові метали). Якщо ж взаємне розташування атомів в рідині не відповідає щільній упаковці (олово, вісмут, германій, кремній), значення R1
обчислене по формулі (93), не співпадає із значенням найкоротшої міжатомної відстані. В цьому випадку експериментальна крива а(S) визначається накладенням ряду кривих, описуваних рівнянням (92).
Співвідношення S1
R1
= 7,73, тобто 4πR1
sinθ = 7,73λ, аналогічно формулі Вульфа-Брегга 2dsinθ = λ. З цих формул виходить, що


Відношення цих величин дає R1
=
1,23d1
.
Рівняння Вульфа — Брегга для цього окремого випадку має вигляд
2R1
sinθ = 1,23λ(94)
Таким чином, параметр S, відповідний першому максимуму а(S), пов'язаний з найкоротшою міжатомною відстанню R1
рівнянням Вульфа—Брегга, в яке введений поправочний коефіцієнт 1,23. Рівняння (94) і еквівалентну йому формулу R1
=
7,73/S1
застосовують у разі молекулярних рідин для оцінки середньої відстані між сусідніми молекулами. При цьому припускають, що перший максимум інтенсивності цілком обумовлюється міжмолекулярним розсіюванням, просторовою конфігурацією молекул і їх упаковкою. Важливо відзначити, що про ступінь ближнього порядку в рідині і твердій аморфній речовині можна судити по ширині і висоті максимумів кривої а(S). Чим більше їх висота, тим менш інтенсивно тепловий рух атомів і тим вищий ступінь їх впорядкованості. Таким чином, маючи експериментальні криві розсіювання, можна по них визначити найкоротшу відстань між атомами і молекулами рідини, з'ясувати характерні особливості розташування найближчих сусідів, тенденції зміни упаковки частинок з температурою. Зв'язок інтерференційної функції із стисливістю. Граничне значення функції а(S) у напрямку до малих кутів розсіювання для будь-якої речовини пов'язано з його стисливістю. При цьому йдеться про граничне значення при S= 0 виразу (88), який був одержаний з точнішого рівняння (87) при виключенні з нього доданку, який визначається зовнішньою поверхнею досліджуваної речовини і не пов'язане з його структурою. Величина а(0), яку визначимо, є граничним значенням розсіюючої здатності речовини, віднесеної до одного атома. Вираз (88) для S =0 перепишемо у вигляді
 (95) (95)
Враховуючи умову нормування функції ρ(R),
одержимо
 
Інтенсивність розсіювання при S = 0 рівна
 (96) (96)
Це співвідношення показує, що значення інтерференційної функції при нульовому куті розсіювання представляється як міра флуктуації числа атомів, що містяться в даному об'ємі. Ці флуктуації пов'язані з коефіцієнтом ізотермічної стисливості βT
= 1/ρat
(dρat
/dp)T
співвідношенням
 (97) (97)
Таким чином,
a(0) = < ρat
>kTβT
(98)
Згідно цій формулі граничне значення а(0) буде більше для речовин (газів), що сильно стискаються, ніж для тих, що малостискаються (рідин, аморфних тіл). Значення а(S) при малих кутах розсіювання різко зростає при підході до критичної точки, що пов'язане з виникненням флуктуації густини — областей згущування і розрідження. Рідина стає все більш «пористою». Безпосередньо біля критичної точки області згущувань чергуються з областями розріджень. Через необмежене зростання стисливості речовини флуктуації густини можуть перевищувати 100 Å. Користуясь формулою S = 2π/d, знаходимо, що розсіювання на флуктуаціях такої величини виявляється при S = 0,06 Å-1
. Це при довжині хвилі λ =1,54 Å відповідає куту розсіювання близько 40`.
Визначивши а(0), можна по формулі (98) обчислити βT
. Проте значення а(0) не можна заміряти експериментально, якщо криві розсіювання виходять від плоскої поверхні зразка. При зйомці на проходження потрібно знати інтенсивність первинного пучка рентгенівського випромінювання або нейтронів. Вимірювання абсолютного значення цієї інтенсивності зв'язане з технічними труднощами. Практично зручніше визначати βT
не через граничне значення інтенсивності а(0), а через радіальну функцію ρ(R).
Замість (95) можна написати
 (99) (99)
звідки
 (100) (100)

Цей вираз вельми важливе для пояснення впливу сил тяжіння і відштовхування на пружні властивості рідин. Приведемо декілька прикладів.
Допустимо, що досліджувана речовина складається з симетричних молекул діаметром а
, між якими діють тільки сили відштовхування. Радіальна функція розподілу, і залежність енергії взаємодії молекул від відстані між ними виглядають так, як показано на мал. 2.12. З нього виходить, що
U(R) = +∞ , ρat
(R) = 0 при R ≤ a(101)
U(R) = 0, ρat
(R) —<ρ> = 0 при R > a
Коефіцієнт ізотермічної стисливості такої системи
 (102) (102)
Оскільки 4/3πa3
— об'єм, що оточує кожну молекулу, в межі якого не може проникнути інша молекула, то (N/V)(4/3)πa3
= 1 і, отже, βT
= 0. Цей результат відповідає моделі твердих непроникних щільно упакованих кульок.
Якщо ті ж молекули притягуються один до одного за законом U(R) = —A/R6
то
 (103) (103)
Перший інтеграл визначає площу під кривою розподілу, що відображає потенційну енергію відштовхування молекул. Її значення рівне одиниці, як і у попередньому випадку. Другий інтеграл визначає площу, яка відповідає області тяжіння молекул. Отже,
 βT > 0 (104) βT > 0 (104)
Цей результат показує, що облік енергії тяжіння молекул приводить до збільшення стисливості речовини, і чим більше ця енергія, тим вище коефіцієнт стисливості.
Переходячи до реальнішої моделі і вважаючи, що взаємодія молекул описується формулою Леннарда—Джонса, одержимо
 (105) (105)
В цьому випадку значення βT
буде ще більше за рахунок збільшення площі, обмеженої кривою розподілу. Таким чином, сили відштовхування молекул зменшують стисливість речовини, сили тяжіння збільшують її. І чим більше крутизна кривої відштовхування, тим менше стисливість і більше пружність. Оскільки функція 4πR2
[ρat
(R) — < ρat
>] перетворюється в нуль на відстані R,
рівному декільком молекулярним діаметрам, то з (100) витікає, що для стисливості рідини визначальне значення має найближче оточення, характер зміни енергії тяжіння і відштовхування молекул на малих відстанях.
Для кількісного опису пружних властивостей рідин потрібні певні значення функції 4πR2
[ρat
(R) — < ρat
>] при малих R, що пов'язане з необхідністю точних вимірювань функції [а(S) — 1] при великих S.
Розрахунки по формулі (100) дають завищені значення βT
якщо функція розподілу має помилкові піки при R < R0
обумовлені обривом кривої при S = Smax
і приблизними значеннями інтенсивності при великих кутах розсіювання. Для усунення помилкових піків слід в інтерференційну функцію ввести множник ехр(—b2
S2
)значення параметра b,
в якому підбирають так, щоб добуток [a(S) — 1] ехр(—b2
S2
)при S = Smax
було рівне приблизно 0,1 свого первинного значення. Проте множення всіх значень інтерференційної функції на ехр(—b2
S2
) приводить до деякого зрушення положення першого і подальших максимумів кривої радіального розподілу. Зв'язок структурного чинника з електронними властивостями металів.
Однією з фізичних властивостей металів, безпосередньо пов'язаних з ближнім порядком і енергією взаємодії частинок, є електропровідність. Розвиток квантової теорії твердого тіла привів до висновку, що електропровідність рідких металів можна обчислити теоретично за експериментальними даними для структурного чинника а(S), задаючи Фурье-образ потенційної енергії взаємодії електронів з атомами розплаву. Основна ідея, на якій базуються розрахунки електропровідності, полягає у тому, що розсіювання електронів провідності рідкого металу описується структурним чинником, аналогічним для рентгенівського випромінювання або нейтронів. Помітимо, що структурний чинник розсіювання електронів провідності обмежений значеннями S, які для одновалентних металів знаходяться зліва від першого максимуму а(S), а для двох (і більш) валентних металів — справа від нього. В той же час, за даними розсіювання повільних нейтронів і рентгенівського випромінювання довжиною хвилі λ
= 0,5—0,7 Å, структурний чинник визначається до S = 15—20 Å-1
. За сучасними уявленнями, електрони провідності металу не можна розглядати як вільні. Їх рух в кристалі модульований періодичним силовим полем гратки. Безперервний енергетичний спектр вільних електронів в просторі розпадається на зони дозволених енергій — зони Бріллюена, розділені інтервалами енергій, забороненими для електронів. На шкалі енергій E(k)зони Бріллюена зображають графічно у вигляді смуг дозволених значень енергії і розривів між ними (мал. 2.13).

У тривимірному k-просторівони мають вид многогранників, форма яких визначається симетрією кристалічної гратки, а розміри — параметрами гратки. Для гранецентрованої кубічної гратки перша зона Бріллюена є октаедром, а для об'ємно-центрованої гратки — кубічний додекаедр. Усередині зони Бріллюена енергія електрона безперервно змінюється з хвильовим числом по параболічному закону
E(k) = h2
k2
/(8π2
m)(106)
Імпульс і хвильовий вектор електрона зв'язані співвідношенням де Бройля р = hk/(2π). У міру наближення хвильового вектора до межі зони енергія електрона відхиляється від параболічної залежності, швидкість його руху сповільнюється, що можна пояснити збільшенням ефективної маси mэф
.
Дійсно, електрон в періодичному полі гратки прискорюється зовнішнім електричним полем, якщо
 (107) (107)
На межі зони d2
E/dk2
перетворюється в нуль, а маса — в нескінченність. Відбувається як би віддзеркалення електронів від площин гратки.
Існування меж зон Бріллюена узгоджується з умовою Вульфа— Брегга для дифракційних максимумів рентгенівського випромінювання. Відомо, що при 2dcosα = mλпучок рентгенівського випромінювання повністю відбивається від площин кристала. Якщо записати цю умову у вигляді |(kn
)| =±πm/d то ми одержимо не що інше, як рівняння площини, що визначає межі зон Бріллюена.
Таким чином, межі зон Бріллюена відповідають таким значенням імпульсів електронів, при яких відбувається дифракція електронних хвиль, що імітують рух електронів провідності металу.
Важлива характеристика енергетичного спектру електронів — ізоенергетична поверхня Фермі, яка в тривимірному k -просторіслужить межею між зайнятими і вакантними рівнями. Тверді тіла, у яких поверхня Фермі проходить в дозволеній зоні, є металами, а тіла, у яких енергетичний спектр складається із заповнених і порожніх зон, — діелектриками або напівпровідниками.
Поверхні Фермі у електронів провідності різних металів складні і не схожі одна на одну. У майже вільних електронів поверхня Фермі сферична. Її радіус визначається по формулі
KF
= (3π2
ρZ)1/3
(108)
Електрони, розташовані поблизу поверхні Фермі, володіють максимальною енергією, званою енергією Фермі. Саме ці електрони обумовлюють електропровідність металу. При русі в розплаві вони розсіваються атомами металу. У боровському наближенні довжина вільного пробігу l
електронів обчислюється з рівняння
 (109) (109)
а питома електропровідність — по формулі
 (109`) (109`)
Тут e —
заряд електрона; nZ — число валентних електронів в одиниці об'єму; S = 2kF
sinθ; φ(S) — Фурье-образ псевдопотенціалу електрон-іонної взаємодії.
Під псевдопотенціалом мається на увазі ефективний періодичний потенціал, що змінює стан руху електронів в розплаві. Електрони провідності відштовхуються від електронних оболонок атомів. Разом з тим вони притягуються до атомних ядер. Різниця між тяжінням і відштовхуванням і представляє розсіюючий потенціал, або псевдопотенціал. Як випливає з (109`), для обчислення питомої електропровідності рідкого металу використовують не всю функцію а(S), а лише ті її значення, які лежать в межах 0 < S <2kF
. Наприклад, для рідкого срібла (Z = 1; ρ = 0,051 ат/ Å3
) k= 1,15 Å-1
.Функція а(S) має межу при S = 2,30 Å -1
, тобто зліва від першого максимуму.
Верхня межа інтеграції в (109) означає, що в рідкому металі зберігається контур поверхні Фермі, усередині якої укладені електрони провідності. Оскільки Sмакс
= 4π/λмин
, то на прикладі срібло видно, що мінімальна довжина хвилі електронів провідності λмин
= 5,46 Å. Якщо б електрони в металі були абсолютно вільні, то їх розсіювання на атомах при русі в зовнішньому електричному полі можна було б спостерігати при тих же значеннях S, що і у разі рентгенівського випромінювання. Досвідом це не підтверджується. Отже, різка верхня межа структурного чинника, що описує розсіювання електронів провідності, пояснюється зонною структурою енергетичного спектру електронів. Можливість обчислення електропровідності рідких металів по значеннях інтерференційної функції і псевдопотенціалу підтверджує наявність прямого зв'язку між структурою і електричними властивостями. На це вперше вказав А. Ф. Іоффе. По думці вченого, процес утворення електронів провідності безпосередньо пов'язаний з ближнім порядком і електронною конфігурацією атомів.
Параметри, визначувані по кривих розподілу атомної густини
З'ясуємо, яку інформацію про структуру рідин і аморфних тіл можна одержати, аналізуючи функцію 4πR2
ρат
(R). Графічно її зображають кривими, що осцилюють щодо 4πR2
<ρат
>. Як приклад приведемо криві радіального розподілу атомів для рідкого олова і аморфного селену (мал. 2.14).

Перша одержана рентгенографічно А.Ф. Ськришевськім, а друга — електрографічно Я.І. Стецивом. Для олова (див. мал. 2.14) крива після першого максимуму не досягає осі абсцис, а на кривий для селену перший максимум дискретний. Нерозв'язність піків функції 4πR2
ρат
(R) відображає наявність в рідині руху трансляції атомів, безперервне переміщення їх з однієї координаційної сфери в іншу і навпаки. Дискретність першого піку функції 4πR2
ρат
(R) є доказом існування фіксованих положень атомів; поступальні переміщення атомів з одних рівноважних положень в інші не спостерігаються. Таким чином, криві функції 4πR2
ρат
(R) для атомарної рідини і твердої аморфної речовини принципово відрізняються тим, що в аморфній речовині перший пік цієї функції розділений проміжком, де 4πR2
ρат
(R) = 0, тоді як в рідині навіть перший пік не визначений з боку великих R. Загальним для рідин і аморфних речовин є розмитість піків радіальної функції атомної густини.Розмитістьїх відбувається унаслідок коливань атомів навколо положень рівноваги і статистичного розкиду центрів коливань.
Положення максимумів на кривій 4πR2
ρат
(R) визначає найвірогідніші відстані, площа під максимумами дає середнє число сусідніх атомів, ширина максимуму на половині його висоти — середньоквадратичне відхилення атомів від рівноважного положення, крива розподілу в цілому характеризує ближній порядок в рідині і аморфній речовині.
Визначення координаційних чисел
. У разі аморфного селену площа першого піку на кривій розподілу при R1
=2,32 Å рівна двом, а другого при R2
= 3,7 Å — восьми, що відповідає числу атомів на даних відстанях. Гратка кристалічного селену складаються із зигзагоподібних гвинтових ланцюжків, кожен атом в яких ковалентно пов'язаний з двома найближчими атомами, а ланцюжки між собою — силами Ван-дер-Ваальса. Відстань між найближчими атомами в ланцюжку рівна 2,34 Å, а між атомами сусідніх ланцюжків — приблизно 3,8 Å. Отже, в аморфному селені зберігається ближній порядок такої ж, як в кристалічному. Неізольованість першого і подальших піків на кривій розподілу для рідкого олова утрудняє вимірювання площі під ними. Кількісно можна інтерпретувати тільки перший максимум функції 4πR2
ρат
(R), обчислити тільки перше координаційне число. При цьому площу під максимумом виділяють двома способами: симетрично, тобто як би дзеркальним відображенням лівої гілки кривої щодо перпендикуляра, опущеного з вершини максимуму на вісь R, інесиметрично — продовженням спадаючої правої гілки кривої до перетину її з віссю абсцис. Перший спосіб заснований на припущенні, що відхилення атомів однакове як у бік збільшення, так і у бік зменшення R щодорівноважного R1
.
Координаційнечисло знаходиться обчисленням інтеграла
 (110) (110)
другий спосіб визначення n1
заснований на припущенні, що перший пік функції 4πR2
ρат
(R) є як би дзеркальним відбиттям кривої залежності потенційної енергії взаємодії атомів від відстані між ними (див. мал. 1.4). У рідинах коливання атомів щодо рівноважних положень ангармонічні. Сили відштовхування з боку центрального атома обмежують зсуви сусідніх атомів від рівноважного положення у бік менших R. Атомы мають набагато більшу свободу руху у бік зростання R щодорівноважної відстані R1.
В результаті в середньому на рівних відстанях від R1
знаходитьсянеоднакове число атомів: при R1
—
R' їхменше, ніж при R1
+
R' що іобумовлює асиметрію першої координаційної сфери щодо R1.
Числоатомів в першій координаційній сфері визначається інтегралом
 (R" > R') (111) (R" > R') (111)
Таким чином, залежно від способу виділення площі першого максимуму кривої розподілу для координаційного числа n1
виходять різні значення. Для рідкого олова по формулі (110) знаходимо n1
= 8,6, а по формулі (111) — n1
= 9,7. У кристалічній гратці олова n1
= 4 + 2 + 4.
Координаційне число, як один із структурних параметрів рідини, пов'язане з взаємодією найближчих сусідів. Значущість цього числа полягає у тому, що воно дозволяє скласти наочне уявлення про характер зміни упаковки при плавленні і подальшому нагріванні розплаву. Проте структура рідини в цілому описується не координаційними числами і радіусами координаційних сфер, а радіальними функціями розподілу. Теоретичні і експериментальні дослідження показують, що координаційне число в рідині є не числом в буквальному розумінні, а своєрідною функцією густини і температури. Координаційні числа мають точні значення лише в кристалі, де функція 4πR2
ρат
(R) дискретна. У рідині вони піддаються флуктуаціям. По теоретичних розрахунках І. 3. Фішера, в рідких металах флуктуація першого координаційного числа n1
складає 10%, а другого n2
—
30—40%. Такі високі значення флуктуації координаційних чисел є слідством руху трансляції атомів разом з коливальним. Найвірогідніше число найближчих сусідів в рідині може не співпадати з середнім його значенням. Тому кількісний опис розподілу найближчих сусідів повинен бути відображене не середнім координаційним числом n1
, афункцією розподілу W(n1
)визначаючої вірогідність виявлення різного числа найближчих сусідів на даній відстані. У простому випадку функція W(n1
)може бути представлена дискретним гаусовим розподілом
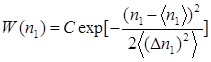   (112) (112)
де n1
, n2
...
— можливі значення першого координаційного числа < n1
>— середньоквадратична флуктуація першого координаційного числа.
На мал. 2.15 показаний розподіл координаційних чисел рідкого аргону і води. Видно, що в рідині окрім середнього координаційного числа n1
можливі інші значення. Наприклад, для води однаково вірогідні n1
= 4 і n1
= 5, значна вірогідність для n1
=
3 і n1
= 6. Функція розподілу W(n1
)насправді не така симетрична, як гаусова. У рідин, порівняно щільно упакованих, переважають флуктуації координаційного числа у бік його зменшення, а у рідин з малою густиною упаковки атомів — у бік його збільшення.
Визначення середньоквадратичного відхилення атомів.Знайдемо зв'язок між середньоквадратичним відхиленням атомів <ΔR2
k
> і шириною піку кривої розподілу на половині його висоти. При цьому вважатимемо, що всі піки мають гаусову форму і однакову ширину (мал. 2.16). Тоді
 (113) (113)
де Rk
—
радіус k-йкоординаційної сфери, nk
— координаційне число, напівширину піку функції 4πR2
ρат
(R) визначимо як
L1/2
= R2
—
R1
(114)
де R1
іR2
— значення R, при яких функція 4πR2
ρат
(R) рівна половині свого максимального значення, тобто коли
 (115) (115)
Замінюючи у формулі (113) функцію 4πR2
ρат
(R) її значенням на половині висоти піку, одержимо рівняння
 (116) (116)
Вирішуючи його, знайдемо
 
Отже,  Тоді шукане середньоквадратичне відхилення атомів в одному напрямі Тоді шукане середньоквадратичне відхилення атомів в одному напрямі
<ΔR2
k
> = 0,18L2
1/2
Оскільки рідини і аморфні тіла ізотропні, той повний квадратичний зсув <ΔR2
> = 3<ΔR2
k
>. Припущення про те, що всі піки функції 4πR2
ρат
(R) мають однакову напівширину, справедливе тільки для кристалів. У разі рідин і аморфних тіл ширина піків цієї функції зростає із збільшенням відстані від фіксованого атома. Згідно Дж. Прінсу, середньоквадратичний зсув атома з рівноважного положення пов'язаний із значенням Rk
співвідношенням
<ΔR2
k
> = 2DRk
(117)
де D — коефіцієнт (що має розмірність довжини), що враховує статистичний розкид в положеннях рівноваги атомів, виникає при плавленні кристала і подальшому підвищенні температури. Його числове значення можна обчислити по формулі, запропонованій В. П. Квітковим,
 (118) (118)
де Q — енергія, необхідна для руйнування кристалічної гратки; Тпл
— температура плавлення; R — молярна газова постійна; Dmax
— максимальне значення D при високій температурі розплаву.
Н. І. Гулівец при теоретичному розгляді залежності <ΔR2
k
> від Rk
в простих рідинах відзначає, що значення <ΔR2
k
> складаються з динамічних зсувів, обумовлених тепловими коливаннями атомів рідини біля рівноважних положень, і статичних зсувів, пов'язаних з розкидом рівноважних положень атомів біля деякого середнього положення. Складові <ΔR2
дин
> і <ΔR2
ст
> описуються формулою
 (119) (119)
де m — маса атома; с — швидкість розповсюдження гіперзвукових хвиль в рідині;
 інтегральний синус; u = hν/(kT). інтегральний синус; u = hν/(kT).
Якщо в простих рідинах динамічні і статичні зсуви атомів відбуваються незалежно, то
<ΔR2
> = <ΔR2
дин
> + <ΔR2
ст
>
Слід зазначити, що експериментальне значення напівширини піку функції 4πR2
ρат
(R) завжди завищене і прагне при зростанні S до істинного по формулі, знайденій Я. І. Стецивом:
 (120) (120)
Наприклад, для рідкого олова крива а(S)була визначена до значення
Smax
= 7,8 Å-
1
.
Істинна напівширина першого піку L1/2
= 0,28 Å.
Монохроматизація випромінювання
Рідини і аморфні тіла на відміну від кристалів не дають дискретних дифракційних максимумів. Тому для дослідження їх структури важливо знати загальний хід інтенсивності залежно від кута розсіювання. Оскільки характеристичний спектр рентгенівського випромінювання складається з дискретних довжин хвиль, кожна з яких дає свою дифракційну картину, то використовуване випромінювання повинне бути монохроматичним. Найінтенсивнішою в рентгенівському спектрі є Kα
-лінія, тому здається природним, що в структурному аналізі рідин використовується саме Kα
- випромінювання. Супроводжуюче його Kβ
- випромінювання розсівається речовиною незалежно від Kα
- випромінювання. В результаті виникають дві дифракційні картини: одна від Kα
-, а інша від Kβ
- випромінювання, що утрудняє їх розшифровку. Тому Kβ
- випромінювання фільтрується. Існує декілька способів монохроматизації рентгенівського випромінювання. Простий з них грунтується на використовуванні селективно-поглинаючих фільтрів. Оскільки довжина хвилі Kβ
- випромінювання менше, ніж Kα
- випромінювання, то можна підібрати речовину, поглинаючу Kβ
- випромінювання сильніше, а Kα
- випромінювання слабкіше. Для цього вибирають елемент, у якого стрибок поглинання λk
знаходиться між двома цими випромінюваннями.
Характеристики фільтрів для Си-, Мо- і Ag-випромінювання, звичайно використовуваних для структурного аналізу рідин, приведені в табл. 3.

Фільтри виготовляють з фольги або шару порошку, закріпленого на папері.
Для поліпшення монохроматизації рентгенівського випромінювання застосовують подвійні (диференціальні) фільтри. При цьому з досліджуваної речовини одержують дві криві інтенсивності: одну з фільтром, межа поглинання якого лежить між Kβ
- і Kα
- випромінюваннями, а іншу з фільтром, що має межу поглинання безпосередньо за Kα
- випромінюванням.

Як видно з мал. 4.1, поглинання Ni- і Co-фільтрів майже однаково для всіх хвиль, крім тих, що знаходяться в інтервалі між 1,487 і 1,607 Å, де Ni-фільтр поглинає слабкіше, ніж Co-фільтр. Якщо джерелом рентгенівського випромінювання є трубка з мідним анодом, то ця смуга включає Kα
- випромінювання довжиною хвилі λ= 1,54 Å і вузьку смужку суцільного спектру щодо слабкої інтенсивності. Якщо криві інтенсивності одержані в однакових умовах, то, віднімаючи з кривої з Ni-фільтром криву з Co-фільтром, одержимо криву, відповідає випромінюванню, близькому до Kα
.
Досконаліша монохроматизація рентгенівського випромінювання досягається відбиванням від монокристалів (кварц, германій, кремній, графіт, фтористий літій). Кристал є пластинкою, одержаною сколюванням по площині спайності кристала. Як відомо, при взаємодії рентгенівського випромінювання з речовиною виникають когерентне, некогерентне і флуоресцентне випромінювання. Якщо довжина хвилі падаючого випромінювання набагато більше довжину хвилі флуоресцентного випромінювання, то останнє можна ослабити відповідним фільтром. Флуоресцентне випромінювання можна майже повністю виключити за допомогою кристала монохроматора, помістивши його за зразком. Якщо реєстратором рентгенівського випромінювання є лічильник, то флуоресцентне випромінювання може бути відфільтроване амплітудним диференціальним дискримінатором. Для виключення некогерентної становлячої розсіювання Б. Уорреном була запропонована методика отримання кривих інтенсивності, заснована на використовуванні флуоресцентного збудження (мал. 4.2).

Вузький потік рентгенівського випромінювання, що виходить з трубки 1 з родієвим антикатодом, після віддзеркалення від кристала 2,
проходячи через щілину S1
падає на зразок 3,
викликаючи когерентне і некогерентне розсіювання. Повне розсіювання проходить через приймальну щілину S2
і потрапляє на молібденовий екран 4,
встановлений під кутом 45° до розсіяного потоку. Когерентна складова розсіяного випромінювання, маючи довжину хвилі λ= 0,615 Å, збуджує в молібденовому екрані флуоресцентне випромінювання довжиною хвилі λ> 0,620 Å, яке реєструється лічильником 5,
розташованим поблизу молібденового екрану. Некогерентне розсіювання має довжину хвилі більше 0,620 Å і, отже, не може порушити в молібдені флуоресцентне випромінювання. Поміщаючи цирконієвий фільтр перед лічильником, можна виключити полікристалічну дифракцію на молібденовому екрані.
Оцінимо порядок довжин хвиль електронів і нейтронів, вживаних в структурному аналізі.
Припустимо, що пучок електронів прискорюється різницею потенціалів U.Швидкість їх руху на шляху від катода до анода визначиться із співвідношення
mυ2
/2 = eU/300 υ = [eU/(150m)]1/2
(121)
а еквівалентна довжина хвилі — по формулі
λ = h/mυ (122)
тобто λ = 10-10
(150/U)1/2
Згідно цій формулі, електронна хвиля порядка 1 Å, відповідна довжині хвилі звичайно застосованого рентгенівського випромінювання, відповідає електронам з прискорюючою напругою 100—150 В. Проте такі електронні пучки майже повністю поглинаються речовиною в декілька атомних шарів. З цієї причини повільні електрони не використовуються для проведення структурних досліджень речовини. Електронографічні дослідження проводять за допомогою електронів, прискорюваних різницею потенціалів в 30—60 кВ, що відповідає довжинам хвиль порядка 0,07—0,05 Å. Так як всі електрони пучка прискорюється однією і тією ж різницею потенціалів, то електронні хвилі можна вважати монохроматичними.
Спектр нейтронів, що виникають в реакторі в результаті ділення ядер урану або плутонію, є, суцільним. Це пояснюється тим, що перед виходом з реактора нейтрони випробовують численні зіткнення з ядрами атомів сповільнювача, розподіл швидкостей яких підкоряється закону Максвела. Відповідно і характер розподілів нейтронів по довжинах нагадує максвеловську криву з максимумом при деякій довжині хвилі λ', визначуваної з умови
mυ2
/2 = (3/2)kT(123)
на підставі якого
λ' = h/mυ = h/(3mkT)1/2
(124)
З цієї формули виходить, що довжини хвиль, відповідні швидкостям нейтронів, що знаходяться в тепловій рівновазі з атомами сповільнювача при температурах 0 і 100°С, рівні відповідно 1,55 і 1,33 Å. Ця обставина вельми важливо, оскільки нейтрони довжиною хвилі такого порядку найбільш зручні для вивчення структури і атомної динаміки твердих тіл і рідин. Метод монохроматизації повільних нейтронів грунтується як на хвильових, так і на корпускулярних властивостях цих частинок. У першому випадку монохроматизація нейтронів виникає при відбиванні від монокристала або полікристалічних фільтрів, в другому — за допомогою механічних переривників.
Для отримання пучка монохроматичних нейтронів на їх шляху при виході з реактора ставлять достатньо великий кристал, що перекриває весь пучок. Оскільки спектр нейтронів суцільний, то будь-якому положенню кристала відповідає деякий інтервал довжин хвиль, для яких кут ковзання задовольняє умові селективного відбивання 2dsinθ = nλ
де n — порядок відбивання. Користуючись формулою
λ = h/(2mE)1/2
(125)
одержимо вираз для кінетичної енергії нейтронів
E = n2
h2
/[2m(2dsinθ)2
] (126)
Звідси видно, що в даному напрямі відбиваються нейтрони з дискретним значенням їх енергії. На практиці використовують відбивання першого порядку. Інтенсивність відбивання n-го порядку в n2
раз слабкіша інтенсивності відбивання першого порядку. Крім того, якщо у відбиванні беруть участь нейтрони з енергією поблизу максимуму спектру, то нейтрони з енергією, що відповідає вищим порядкам відбивання, потраплятимуть в інтервал спаду кривої максвеловського розподілу, що також обумовлюється зменшенням відносної інтенсивності відбивань вищих порядків. У якості монохроматорів використовуються монокристали свинцю, міді, цинку, берилію, германію, характерними властивостями яких є велике значення амплітуди когерентного розсіювання при малому поглинанні. Повертаючи кристал на певний кут, можна виділити з суцільного спектру нейтронів вузьку смужку довжин хвиль вширшки порядка 0,05 Å.
Дія кристалічних фільтрів, використовуваних для монохроматизації нейтронів, заснована на тому, що нейтрони з довжиною хвилі λ< 2dmax
зазнають відбивання від системи різноманітно орієнтованих площин кристала і тим самим виводяться з пучка. Через фільтр проходять лише ті нейтрони, довжини хвиль яких λ> λ
гр
,де λ
гр
— гранична довжина хвилі для даної речовини. Метод фільтру забезпечує велику інтенсивність пучка, проте не дає достатній монохроматичності.
Принцип механічної монохроматизації полягає в розділенні нейтронів по швидкостях, точніше, по різниці часів, що вимагаються для прольоту заданої відстані. Наприклад, сфазовані диски, що обертаються, з щілинами, розташованими на певній відстані одна від одної, пропускатимуть нейтрони тільки певної енергії, створюючи імпульсний пучок квазімонохроматичних нейтронів. Відзначимо особливості методу електронографії. Він істотно відрізняється від рентгено- і нейтронографічного методів тим, що інтенсивність розсіювання електронів атомом майже в 106
разів перевищує інтенсивність розсіювання рентгенівського випромінювання і нейтронів. Це обумовлює швидкість проведення електронографічних досліджень і його незамінність при вивченні будови молекул газів, структури тонких плівок і кінетики їх виникнення. Разом з тим електронографія поступається рентгено- і нейтронографії діапазоном практичного застосування. Електрони сильно поглинаються речовиною. Тому електронограми можна одержувати тільки від тонких шарів зразка. Зйомка з вільної поверхні рідини практично неоможлива, тому що зразок, опромінюваний електронами, повинен поміщатися всередину електронографа, з якого повітря відкачане до тиску порядка 10-4
Па. Застосовувати герметичні кювети з досліджуваною рідиною не можна через сильне поглинання пучка електронів їх стінками. Метод електронографії застосовний переважно до твердих тіл і газів.
Є труднощі при визначенні інтенсивності когерентного розсіювання електронів. Досвід показує, що інтенсивність розсіювання електронів швидко убуває із зростанням кута розсіювання, досягаючи вже при S ≈ 7 Å-1
дуже малих значень. Це утрудняє вимірювання інтенсивності дальнекутового розсіювання електронів, що містить інформацію про міжатомні відстані в досліджуваній речовині, про кількість найближчих сусідів і особливо про середні відхилення атомів від рівноважного положення. З метою посилення дальніх дифракційних максимумів було запропоновано вимірювати не I(S), а I(S)/f2
(S), що легко здійснити за допомогою сектора, що обертається. Він є пристроєм з одного або двох металевих пелюсток серцеподібної форми і поміщається безпосередньо перед фотопластиною, реєструючу дифракційну картину. Під час отримання електронограми сектор приводиться в обертання, чим досягається різний час експозиції для малокутової і дальнекутової частин дифракційної картини. Частота обертання сектора 800—1000 с-1
. Звичайно використовують сектори, форма вирізу яких задається рівняннями r3
= R3
φ/360 (для однопелюсткових) і r3
= R3
φ/180 (для двохпелюсткових), де φ— полярний кут; R — максимальне значення радіусу r сектора. Основна задача сектора — зменшити швидкий спад інтенсивності від центру пластинки до периферії і тим самим в десятки разів підвищити точність вимірювань інтенсивності дальнекутового розсіювання електронів. Секторна методика успішно застосована І. Д. Набітовічем і Я. І. Стецивом в Львівському політехнічному інституті при проведенні досліджень структури аморфних і кристалічних плівок; Л.В. Вілковим, В. П. Спірідоновим і іншими в Московському університеті при дослідженні будови газових молекул. Важливе те, що на електронограмах, знятих за допомогою секторів, що обертаються, немає великої відмінності в густині почорніння в її центральній і периферійній частинах, унаслідок чого часто відпадає необхідність обліку поправки на нелінійність залежності почорніння від експозиції.
Таким чином, секторна методика дозволяє виключити швидко спадаючий фон, викликаний некогерентним розсіюванням, розширити кутову область вимірюваної інтенсивності і тим самим збільшити інформацію про структуру речовини. Наприклад, якщо на електронограмах, знятих без сектора, виявляються лише три дифракційні максимуми до S ≈ 6 Å-1
, то на електронограмах тієї ж речовини, знятих за допомогою сектора, видно десять максимумів до S ≈ 23 Å-1
(мал. 4.3).

Реєстрація розсіяного випромінювання
Для реєстрації рентгенівського випромінювання часто використовують спеціальну рентгенівську фотоплівку. У разі, коли необхідні прямі кількісні вимірювання інтенсивності рентгенівського випромінювання, застосовують гейгерівські і сцинтиляційні лічильники. У основі фотографічного способу реєстрації рентгенівського випромінювання і електронів лежить фотохімічна дія їх на фотоемульсію. В результаті фотохімічного процесу відбувається розкладання молекул AgBr в емульсивному шарі і утворення дрібних зерен срібла. При прояві ці зерна укрупнюються, одночасно відбувається подальше розкладання бромистого срібла на засвічених ділянках плівки. При фіксації зерна, що не розклалися, видаляють з емульсії, а непрозорі зерна металевого срібла залишаються, викликаючи почорніння плівки. Інтенсивність розсіяного випромінювання пропорційна почорнінню плівки, яке вимірюється за допомогою мікрофотометрії. При цьому почорніння в даній точці плівки визначають як логарифм відношення інтенсивності падаючого на плівку світла до інтенсивності світла, що пройшло крізь неї:
B = ln(I0
/I) (127)
Перевагою фотографічного методу є те, що розсіяне під всілякими кутами проміння фіксується на плівці одночасно. До числа недоліків слід віднести тривалість експозиції, важко контрольовані процеси прояву, наявність вуалі і необхідність фотометрування плівки. Це, проте, не знижує значущості фотографічного методу, який і в даний час широко використовують при аналізі структури металів, мінералів, орієнтованих полімерів. Метод реєстрації рентгенівського випромінювання лічильником Гейгера заснований на явищі іонізації молекул газу, тобто утворенню в лічильнику позитивно заряджених іонів і електронів під дією фотонів рентгенівського випромінювання. Гейгеровській лічильник є наповненим газом циліндром з дротяним анодом, розташованим по осі циліндра. До електродів лічильника прикладена постійна напруга, при якому виникає самостійний електричний розряд. При попаданні рентгенівського фотона в лічильник вибивається електрон з атома газу, що наповнює лічильник. Кожен електрон на своєму шляху від місця виникнення до анода в свою чергу викликає лавину електронів, причому кожна лавина протікає незалежно від іншої лавини, викликаючи розряди, реєстровані як електричні імпульси. Чутливість лічильника до рентгенівського випромінювання залежить від його параметрів. Основними з них є ефективність, що дозволяє здатність, рахункова характеристика і власний фон лічильника.
Оскільки в газах рентгенівське випромінювання поглинається слабо, лічильник Гейгера реєструє лише невелике число минулих через нього фотонів. Тому ефективність газонаповнених лічильників до цього випромінювання невелика.
Ефективнішими для рентгеноструктурних досліджень рідин є сцинтиляційні лічильники. Вони є поєднанням: а) кристала-сцинтилятора йодного натрію, активованого талієм, б) фотоелектронного помножувача (ФЕП); в) попереднього підсилювача на транзисторах. Кристал має циліндрову форму з діаметром 20 мм і завтовшки 1 мм. Він герметично упакований в світлонепроникну оправу з тонким берилієвим вікном і встановлюється на фотокатод ФЕП. Оптичний контакт кристала з ФЕП створюється за допомогою силіконового масла. Для поліпшення світлозбираня на задню стінку кристала нанесений алюміній завтовшки близько 10-3мм.
У основі дії сцинтиляційного лічильника лежить здатність кристала NaI (Tl) випускати світлові спалахи під дією заряджених частинок або рентгенівського випромінювання. За допомогою фотоелектронного помножувача ці спалахи перетворяться в електричні імпульси. Процес утворення електричного імпульсу, відповідного енергії фотона падаючого випромінювання, відбувається таким чином. Фотони рентгенівського випромінювання, проникаючи в сцинтилятор, втрачають свою енергію на іонізацію, збудження і частково на дисоціацію молекул. Частина цієї енергії перетвориться в енергію випромінювання—сцинтиляції. Фотони сцинтиляцій, потрапляючи на катод ФЕП, вибивають з нього електрони, кожний з яких, швидшаючи в електричному полі на шляху до першого діноду, одержує енергію, достатню для того, щоб вибити з нього n електронів. Цей процес, розвиваючись лавиноподібно від дінода до діноду, створює на виході ФЕП електричний імпульс, пропорційний кількості електронів, вибитих з фотокатода. З виходу ФЕП імпульс подається на підсилювач, а потім на дискримінатор, який виділяє зі всього спектру імпульсів тільки ті, амплітуда яких відповідає енергії когерентно розсіяних рентгенівських фотонів.

На мал. 4.4 показана схема установки для дослідження структури рідин. Пучок рентгенівського проміння, що вийшов з трубки 1, після формування в коліматорі S1
прямує на циліндровий зразок 2 рідини. Минулий крізь нього первинний пучок поглинається пасткою 3.
На шляху розсіяних променів знаходиться кристал 4,
який відбиває Kα
-випромінювання, реєстроване сцинтіляційним лічильником 5. Розташування монохроматора після зразка дозволяє звести до мінімуму попадання в лічильник флуоресцентного випромінювання. Для отримання картини розсіювання від плоского зразка застосовує θ—θ- дифрактометр. Його особливість полягає у тому, що в процесі зйомки відбувається обертання рентгенівської трубки і лічильника назустріч один одному навколо осі, що проходить через точку зіткнення рентгенівського променя з поверхнею зразка. При цьому кут, під яким випромінювання падає на поверхню зразка, зберігається рівним половині кута розсіювання. Тим самим виключається чинник абсорбції, оскільки він не залежить від кута розсіювання. У сучасній рентгенівській апаратурі для вимірювання кутового розподілу інтенсивності розсіяного випромінювання застосовують дифрактометри, забезпечені сцинтиляційними лічильниками і рахунково-вирішальними пристроями.
Відзначимо, що найважливішою характеристикою сцинтиляційного лічильника і всієї реєструючої апаратури є дискримінаційні криві, які показують залежність кількості зареєстрованих імпульсів від початкового порогу дискримінації при ширині вікна дискримінації 1 В. Форма дискримінаційних кривих залежить від спектрального складу рентгенівського випромінювання, що направляється на сцинтилятор, напруги на фотопомножувачі і коефіцієнта посилення. Незмінність за часом дискримінаційної кривої залежить від стабільності роботи всього комплексу рентгенівської апаратури. При правильному виборі режимів, роботи лічильника амплітудний розподіл реєстрованих імпульсів монохроматичного випромінювання має вигляд, показаний на мал. 4.5.

Накривійповиннібутичітковидношумовачастинаіпікмаксимальноїамплітудиреєстрованоговипромінювання. ПодібнукривуамплітудногорозподілуможнаодержатишляхомпідборунапругинаФЕПікоефіцієнтапідсилення. Відсікаючипорогомдискримінаціїшумилічильникаівстановившипотрібнуширинувікнадискримінації, можнадобиватисявисокоїмонохроматизації реєстрованихімпульсів. ВідповіднодоприведеноїкривоїдляCu-випромінювання значенняпорогудискримінаціїнамаксимумікривоїповиннебутирівне 24 В. Обравширинувікнарівної 18 В, одержимоумовидискримінації, приякихпрактичновесьпікдискримінаційноїкривоїреєструватиметьсярахувальнимпристроєм, забезпечуючитимсамимдостатнійрівеньмонохроматізациіівисокуінтенсивність. ВідношенняΔU (ширинадискримінаційноїкривоїнаполовинівисоти) доUmax
характеризуєамплітуднийрозподілреєструючоїапаратуриідлямідноговипромінюванняскладаєблизько 50 %.
Важливою умовою ефективності лічильника є ширина його вхідної щілини. Вона повинна бути такою, щоб не розмивати істинний профіль дифракційних максимумів і в той же час забезпечувати достатньо реєстровану інтенсивність розсіяного випромінювання. Звично ширину приймальної щілини лічильника і щілини коліматора вибирають приблизно рівною.
Реєстрація розсіяних електронів здійснюється за допомогою фотопластин, а нейтронів — за допомогою лічильників, наповнених трьохфтористим бором. Ядра бору сильно поглинають нейтрони. Захопивши нейтрон, ядро бору перетворюється на ядро літію з випуском α – випромінювання:
1
0
n + 10
5
B→7
3
Li + 4
2
He
Альфа-випромінювання реєструється лічильником по іонізації газу.
Зразки для дослідження
Для отримання кривих інтенсивності від рідини з малим коефіцієнтом поглинання рентгенівського випромінювання застосовують циліндрові зразки. Вони є трубками з пірексового скла завтовшки стінки не більш 0,01—0,03 мм, наповнені досліджуваною рідиною і ретельно запаяні з обох кінців. Замість скляних трубок використовують кювети з дуже тонкими плоско паралельними віконцями.
Оскільки в цих випадках рентгенограми виходять в проходячому промінні, необхідно брати зразок оптимальної товщини, оскільки дуже тонкий шар містить недосить розсіюючої речовини, а в товстому шарі відбувається велике поглинання.
Оптимальна товщина шаруючи речовини, що бере участь в розсіянні рентгенівського випромінювання, рівна зворотному значенню коефіцієнта лінійного ослаблення:
l
0
= 1/μ(128)
де 
Тут ρ— густина речовини; М — молярна маса; μ/ρi
— масовий коефіцієнт ослаблення i-го атома; Ai
— його атомна маса; ni
— число атомів i-го сорту.
При дослідженні рідин з великим, коефіцієнтом поглинання рентгенограми виходять відбиттям від вільної поверхні зразка. В цьому випадку дифракційна картина фіксується з тієї сторони від поверхні рідини, з якою на неї прямує первинний потік рентгенівського випромінювання. При цьому кут між первинним пучком і горизонтальною поверхнею зразка не повинен перевищувати 8—10°, інакше інтерференційні максимуми можуть виявитися у області геометричної тіні зразка.
Якщо вимагається одержати рентгенограму від рідини при високих температурах (метали), її поміщають в тигель з тугоплавкого матеріалу, що хімічно не взаємодіє з розплавленим металом. Нагрів зразка здійснюється електричним струмом. Для усунення можливості окислення зразка камеру наповнюють гелієм. При дослідженні рідин методом дифракції нейтронів застосовують зразки у вигляді кварцових, алюмінієвих або ванадієвих ампул, заповнених досліджуваною речовиною і ретельно запаяних з обох кінців. Діаметр зразка залежить від поглинаючої здатності рідини. Звично він порядка 10—15 мм. Зразки для отримання електронограм рідин і твердих аморфних речовин є плівками завтовшки 300—500 Å.
Внесення поправок на поляризацію і поглинання
Рентгенівське випромінювання при розсіянні речовиною частково поляризується, унаслідок чого ослабляється його інтенсивність. Разом з тим необхідно знайти інтенсивність, яка спостерігалася б за відсутності поляризації.
Якщо крива інтенсивності одержана у фільтрованому випромінюванні, то поправка на поляризацію обчислюється по формулі
P(θ) = (1 + cos2
2θ)/2 (129)
де 2θ — кут розсіювання.
Якщо ж первинний потік рентгенівського випромінювання монохроматизується при відбиванні від монокристала, то формула для обчислення поправки на поляризацію має вигляд
P(θ) = (1 + cos2
2θ cos2
2φ)/2 (130)
де φ — кут відбивання від відповідної площини монокристала. Поляризаційний чинник для нейтронів і електронів при їх розсіянні рівний одиниці.
При взаємодії рентгенівського випромінювання, електронів і нейтронів з речовиною частина їх енергії перетворюється на різні види внутрішньої енергії речовини і в енергію вторинного випромінювання. Це приводить до часткового поглинання падаючого на зразок випромінювання. Тому інтенсивність розсіювання не може бути правильно визначена без внесення поправки на поглинання. Ця поправка залежить від форми зразка і кута розсіювання. У разі плоского зразка при зйомці на проходження проміння ця поправка обчислюється по формулі
 (131) (131)
де l
— товщина зразка; μ — лінійний коефіцієнт поглинання; x = (1—cos2
θ)/cos(2θ)
При зйомці на відбивання від плоскої поверхні зразка поправка на поглинання задається формулою
 (132) (132)
де α — кут, під яким випромінювання падає на поверхню рідини.
Якщо під час зйомки на θ—θ-дифрактометрах зберігається постійність кутів (α = θ), то з (132) витікає, що А не залежить від кута розсіювання А =
1/(4μ).
У разі циліндрового зразка поправка на поглинання може бути розрахована по формулі
 (133) (133)
де R0
— радіус зразка; an
— коефіцієнт, залежний від кута розсіювання.
Значення A(θ) протабульовано для різних μR0
.
Знаючи з умов експерименту μR0
знаходимо за табличними даними A(θ). Внесення вказаних поправок можливо зробити при діленні експериментальних значень інтенсивності на добуток чинників поляризації і поглинання.
Нормування кривих інтенсивності
У рівняннях для інтенсивності розсіювання величини I(S) іF2
(S)виражені в електронних одиницях. З експерименту ми одержуємо інтенсивність у відносних одиницях. Тому необхідно нормувати експериментальні значення інтенсивності, тобто приводити їх до електронних одиниць:
Iнорм
(S) = k Iотн
(S) (134)
Нормуючий множник k
може бути знайдений декількома способами. Один з них заснований на тому, що при великих кутах розсіювання крива інтенсивності перестає осцилювати щодо кривої незалежного розсіювання. Це слідує, зокрема, з рівняння
 (135) (135)
При S→ ∞ функція sinSR/(SR) → 0, а I(S) → NF2
(S).Тому для тих значень S,
при яких міжатомні інтерференційні ефекти виражені дуже слабо, експериментальну криву інтенсивності, виправлену на поляризацію і поглинання, можна сумістити з кривою незалежного розсіювання, розрахованої за табличними даними. Оскільки заміряна інтенсивність складається з когерентної і некогерентної частин, нормуючий множник слід обчислювати по формулах:
 (136) (136)
— для атомарних рідин,
 (137) (137)
— для молекулярних рідин.
Якщо всі експериментальні значення інтенсивності помножити на нормуючий множник, ми одержимо криву розсіювання в електронних одиницях. Після нормування I(S)з неї слід відняти інтенсивність некогерентного розсіювання.
Інший спосіб нормування експериментальних кривих розсіювання заснований на законі збереження інтенсивності, який можна сформулювати так: інтенсивність розсіювання не залежить від того, як розташовані атоми один щодо одного, чи утворюють вони кристал, молекули рідини або газу. Інтерференція між хвилями, розсіяними даним числом атомів, приводить лише до перерозподілу інтенсивності, посиленню в одних напрямах і ослабленню в інших, не змінюючи сумарної її величини. Тому якщо нормовані експериментальні значення інтенсивності проінтегрувати по всіх S,
то цей інтеграл буде рівний інтегралу по значеннях інтенсивності, що дається ізольованими атомами:
 (138) (138)
Експериментальна інтенсивність складається з когерентної і некогерентної частин. Тому рівність (138) слід переписати у вигляді
 (139) (139)
звідки
 (140) (140)
Згідно цій формулі, нормуючий множник визначається як відношення площі під кривою сумарного незалежного розсіювання до площі під експериментальною кривою розсіювання. Критерієм точності нормування може служити рівність
 (141) (141)
яке виходить з рівняння
 (142) (142)
За умови, що для значення R= 0 функція ρ(R) = 0 ,asinSR/(SR) =
1. Средню атомну густину обчислюють по формулі
 або або  (143) (143)
де А — атомна маса, ρ —густина речовини, NA
— постійна Авогадро, mH
— маса атома водню. Середня електронна густина речовини рівна
 (144) (144)
де Zj — число електронів атома.
Необхідно відзначити, що на досвіді криву інтенсивності I(S)можна визначити в обмеженому інтервалі S, а не від 0 до ∞, як це потрібне теорією. Пояснюється це двома причинами: 1) при зйомці на проходження розсіяне під невеликими кутами випромінювання перекривається первинним пучком, а при зйомці на відбивання —краями зразка. Через це не можна визначити хід інтенсивності від 0 до деякого значення S1
Тому доводиться довільно екстраполювати I(S)до нуля; 2) в результаті кінцівки довжини хвилі крива I(S)може бути визначена до значень
S2
< (4π/λ)sinθ. Якщоλ = 1,54 Å, те граничне значення S2
=(4π/λ)=8,1 Å-1
і S2
= 18 Å-1
при λ= 0,71 Å. Експерементально вдається знайти осциляції I(S) до значень S2
= 10 — 12 Å-1
залежно від чутливості методу і довжини хвилі використовуваного випромінювання.
Розглянуті способи нормування експериментальних кривих інтенсивності відносяться до рентгенографії. Нормування кривих розсіювання електронів ускладнюється через відсутність функції некогерентного розсіювання. Ослаблення некогерентного фону за допомогою електронних фільтрів не завжди забезпечує необхідну точність визначення структурних параметрів досліджуваних речовин по їх електронограмам.
І. Д. Набітовіч, Я. І. Стецив і Я В. Волощук запропонували новий метод визначення когерентної інтенсивності і інтенсивності фону по експериментальній кривій розсіювання електронів. Розглянемо суть цього методу. Відомо, що експериментально заміряна інтенсивність розсіювання електронів включає некогерентний фон. Отже,
Iэкс
(S) = Iк
(S) + Iф
(S) (145)
Відповідно до закону збереження інтенсивності когерентна частина нормується за допомогою рівності
 (146) (146)
Інтегруючи (145) і враховуючи Ik
(S),
одержимо
 (147) (147)
де
 (148) (148)
— інтенсивність, яка виходила б в аналогічних умовах від незалежних атомів. За визначенням,
a(S) — 1 = [Iнор
(S) — f2
(S)]/f2
(S) (149)
Враховуючи, що Iнор
(S) = kIk
(S)і беручи до уваги рівність (145) і (148), знайдемо
a(S) — 1 = k[Iэкс
(S)/f2
(S) — <I(S)>/f2
(S)] (150)
Рівняння для розрахунку функції 4πR2
ρ(R) має вигляд
 (151) (151)
У ньому невідомими є нормуючий множник kі доданок <I(S)>/f2
(S). Як показують дослідження, значення функції розподілу сильно залежать від нормуючого множника k, тому виникає питання, як його визначити. З (147) витікає, що крива Iэкс
(S) повинна осцилювати навколо кривої <I(S)>. Отже, крива Iэкс
(S)/f2
(S) також повинна осцилювати навколо кривої <I(S)>/f2
(S). Хід цієї кривої можна визначити графічно. Для цього за експериментальними даними слід побудувати графік функції Iэкс
(S)/f2
(S) залежно від S.
Потім провести криву <I(S)>/f2
(S) так, щоб виконувалася умова
 (152) (152)
При цьому верхню межу інтеграції бажано брати як можна велику, використовуючи тим самим всі спостережувані інтерференційні ефекти.
Щоб знайти нормуючий множник, потрібно знати інтенсивність когерентного розсіювання і інтенсивність фону. З рівнянь (149) і (150) знаходимо
Ik
нор
(S) = f2(S){k[Iэкс
(S)/f2
(S) — <I(S)>/f2
(S)] + 1} (153)
Аналогічно, користуючись рівністю (148), визначимо
Iф
нор
(S) = kIф
(S) = f2
(S)[k I(S)>/f2
(S) — 1] (154)
Теоретичні розрахунки показують, що значення нормуючого множника залежать від верхньої межі інтеграції в рівнянні (151). Межі можливих значень kможуть бути визначені по експериментальній кривій інтенсивності. Як вже відомо, інтенсивність когерентного розсіювання є величиною позитивною, отже,


Ця нерівність показує, що нижня межа параметра 1/kможе бути визначена по значенню найглибшого мінімуму на кривій Iэкс
(S)/f2
(S) тобто
1/kmin
= [<I(S)>/f2
(S) — Iэкс
(S)/f2
(S) ]max
(155)
Інтенсивність фону — теж позитивна величина. Тоді
 тобто тобто 
Верхня межа параметра 1/kвизначиться якнайменшим значенням функції <I(S)>/f2
(S) ,тобто
 (156) (156)
Нерівності (155) і (156) обмежують можливі значення 1/k.
Як нормуючий множник можна узяти середнє значення, обчислене із співвідношення
 (157) (157)
На мал. 4.6 як ілюстрація показані криві Iэкс
(S)/f2
(S) і <I(S)>/f2
(S) для знаходження 1/kmin
і 1/kmax
. Згідно малюнку якнайменше значення <I(S)>/f2
(S) = 4,3 при S = 1,5 Å-1
, а найбільше значення різниці [<I(S)>/f2
(S) — Iэкс
(S)/f2
(S) ]max
= 2,5 при S =4,0 Å-1
. Отже, 1/k= (4,3 + 1,5)/2 = 2,9; k= 0,35. Висловлений спосіб визначення нормуючого множника і інтерференційної функції розсіювання електронів не пов'язаний з громіздкими обчисленнями. Він простий і доступний. На прикладі германію і кремнію було показано, що визначувані цим методом структурні параметри повністю співпадають зданими рентгенографічних досліджень.

Точність визначення структурних параметрів
Як наголошувалося, основними кількісними характеристиками структури рідин є радіальні функції розподілу атомної і електронної густини.
Точність, з якою можуть бути визначені міжатомні відстані і числа найближчих сусідів, зв'язана: а) з наближеним характером рівнянь, що зв'язують структуру речовини з кутовим розподілом інтенсивності розсіювання, обмеженою точністю табличних значень атомних чинників і некогерентного розсіювання, неоднозначністю вибору нормуючого множника; б) з труднощами експериментального характеру (наприклад, обривом кривої інтенсивності при кінцевому значенні S), а також неточностями вимірювання і обліку різних чинників; погрішностями визначення коефіцієнта поглинання, впливом некогерентного фону. Подолання експериментальних труднощів досягається зйомкою в строго монохроматичному випромінюванні, застосуванням сцинтиляційних лічильників для реєстрації розсіяного рентгенівського випромінювання, секторної методики в електронографії. Сучасна апаратура дозволяє вимірювати інтенсивність розсіювання з точністю 2—3%. Вплив обриву кривої інтенсивності на вигляд функції розподілу піддається аналітичному опису. Всесторонній аналіз цього питання був проведений В. Н. Пилиповичем, Р. Хоземаном, Я. І. Стецивом і ін.
Помилкові максимуми радіальних функцій розподілу
. Найістотнішою у визначенні структурних параметрів рідин і аморфних тіл є помилка, що виникає через обрив кривої інтенсивності. Вона може привести до виникнення помилкових максимумів радіальної функції розподілу, до зміни положення максимумів, їх ширини і форми. Щоб виробити кількісну оцінку цієї помилки, потрібно знати функцію а(S)для явно відомого розподілу атомів. З цією метою скористаємося рівнянням (135), з якого виходить, що
 (158) (158)
Виключаючи нульове розсіювання, одержимо
 (159) (159)
Припустимо, що максимуми на кривій розподілу атомної густини мають форму кривих Гауса. Тоді загальна функція може бути представлена у вигляді
 (160) (160)
Підставляючи (160) в (159) і обчислюючи інтеграл, знаходимо
 (161) (161)
З цього рівняння виходить, що чим більше <ΔRk
2
> тим швидше затухає а(S), осцилюючи щодо одиниці. Якщо ж <ΔRk
2
> = 0, що відповідає розподілу атомів в кристалі, то
 (162) (162)
Скориставшися аналітичним виразом а(S), знайдемо функцію розподілу, відповідну k-му координаційному максимуму, по формулі
 (163) (163)
де Smax
— найбільше значення S, до якого визначена крива а(S);Rk
— відстань від початку координат до основи перпендикуляра, опущеного з вершини максимуму кривої розподілу на вісь абсцис.
Цей інтеграл обчислюється аналітично тільки для двох граничних випадків: Smax
→ ∞ і Smax
→ 0. У першому випадку одержуємо початкову функцію Гауса:
 (164) (164)
Коли Smax
мало, другим доданкам у фігурних дужках (163) можна знехтувати. Поклавши
  , знайдемо , знайдемо
 (165) (165)
Оскільки у області координаційного максимуму другий член малий порівнянню з першим, то
 (166) (166)
Цей результат показує, що якщо експериментальну криву інтенсивності обмежити малими значеннями Smax
то на кривій радіального розподілу атомів окрім піку при R= Rk
, відповідного k-йкоординаційній сфері, з'являється ряд побічних (помилкових) піків, що тягнуться в область розташування сусідніх піків. Помилкові максимуми розташовуються майже симетрично по обидві сторони від центрального максимуму. Їх положення знаходиться по формулі
R = Rk
±2,5π/ Smax
(167)

На мал. 4.8 приведені нормована крива інтенсивності і крива 4πR2
ρ(R) одержані для рідкої ртуті А. Ф. Ськришевськім сумісне з Д. П. Карлікової і Д. Н. Карликовим. Вимірювання інтенсивності проведені до значення sinθ/λ = 0,62 Å-1
що відповідає Smax
= 7,8 Å-1
. Поява помилкових максимумів по обидві сторони від істинних на кривій 4πR2
ρ(R) можна чекати при 2,1 і 4,13 Å для R1
= 3,12 Å і при 5,2 і 7,1 Å для R2
= 6,2 Å.
Як видно з малюнка, побічні максимуми відповідають приблизно цим же відстаням. Помилкові максимуми функції 4πR2
ρ(R) з періодом ΔR =±2,5π/ Smax
можуть з'явитися через неточність вимірювання інтенсивності при великих кутах розсіювання. Це завжди треба мати на увазі, оскільки помилки вимірювання I(S)після множення на S можуть істотно спотворити результати.
Амплітуда помилкових піків функції радіального розподілу помітно зменшується при множенні функції S[а(S) — 1] на чинник ехр(— bS2
).Коефіцієнт b вибирають з умови ехр(— bS2
) ≤ 0,1. Наприклад, для рідкої ртуті при Smax
= 7,8 Å-1
знаходимо b =
0,038 Å2
.
Обчислення координаційних чисел
Координаційне число визначається за площею під максимумом, яка у разі одноатомної речовини аналітично представляється формулою
 (168) (168)
Якщо для обчислення цього інтеграла вибрати вираз (160), то набудемо істинне значення координаційного числа
 (169) (169)
Якщо ж скористатися виразом (166), то
 (170) (170)
Межі інтеграції визначаємо з умови (мал. 4.9)
sinSmax
(R—Rk
) = 0 , Smax
(R—Rk
) = ± π , R = Rk
± π/Smax
Таким чином, координаційне число, обчислюване за площею під максимумом теоретичної кривої розподілу, знаходиться в межах
nk
≤ n ≤ 0,18nk
(171)
Тут проаналізовані далеко не всі джерела погрішностей у визначенні функції 4πR2
ρ(R). Але вже з сказанного ясно, що за допомогою дифракційних методів достовірний результат може бути одержаний лише на основі ретельно проведеного експерименту і обліку всіх вірогідних погрішностей обчислень. Ознаками правильного визначення функції розподілу є: а) незначна осциляція кривої 4πR2
ρ(R) при малих R; б) відсутність мінімумів нижче осі абсцис і малих максимумів, розташованих симетрично по обидві сторони від головних максимумів. Помилкові максимуми на кривих розподілу, які можуть з'явитися при розрахунку, ускладнюють визначення істинної картини будови рідини або аморфної речовини. Тому при дослідженні структури не слід обмежуватися тільки кривими розподілу, необхідно детально аналізувати також і криві інтенсивності, оскільки вони безпосередньо пов'язані з розташуванням атомів і молекул.






Методика роботи та аналіз результатів
1.Формування зразків
2.Отримання дифрактограм зразків
3.Побудова радіальних функцій
4.Визначення відстаней та координаційних чисел
1.Зразки формувалися з Мо у вигляді тонких плівок утворених за допомогою ионо-плазменого розпилення. Формування плівок в цих умовах створює наднерівноважні умови і це приводить до того що Мо знаходиться в наднерівноважному стані. Тому ці зразки являються вихідними зразками. По політермах були визначенні ключеві температури при яких суттєво змінювался електричний опір плівок тобто відбувалися фазові переходи при яких змінювалася і їх структура. Таких температур було визначено дві: 600 ºС та 700 ºС. Тому шляхом відпалу цих зразків були отримані зразки відповідно при відпалах 600 ºС та 700 ºС.
2.Дифрактограми отримувалися скануванням рентгенограм отриманих шляхом зйомки зразків в камерах РКД на рентгенівських установках УРС-60.
3. По отриманим дифрактограмам були визначені залежності інтенсивності від кута; і ці значення були занесені в спеціальну програму середовища Excelза допомогою якої були внесені необхідні поправки і побудована радіальна функція густини.
4. З побудованих радіальних функцій в середовище Excel були визначенні відстані та координаційні числа шляхом заміни кривої радіальної функції сумою чотирьох гаусовських кривих. Для вихідного Мо координаційні числа на відстанях першої, 3-ї та 4-ї координаційних сфер дуже близькі до кристалічного Мо, а координаційне число 2-ї координаційної сфери значно менше ніж в кристалічному. Таким чином були встановлені відстані для чотирьох координаційних сфер. При відпалах відстані координаційних сфер спочатку збільшуються, а потім зменшуються але не суттєво; але при 700 ºС третя координаційна сфера по суті має дві відстані: 4,2 Å та 4,54 Å, що свідчать про початок суттєвої перебудови структури.
Висновок
Структурні зміни Мо при відпалах до 700 ºС приводять до формування нанокристалічного стану.
|
