После отбора клонированных рекомбинантных молекул нужно охарактеризовать содержащуюся в них вставку. Для этого необходимо ответить на несколько вопросов. Прежде всего - действительно ли рекомбинантная молекула содержит нужный сегмент ДНК? Далее - включился ли сегмент целиком или только частично? Соответствует ли он во всех деталях геномной ДНК, из которой происходит? Является ли нужная последовательность функциональной? Мы рассмотрим разные подходы к характеризации клонированных фрагментов ДНК. Прежде всего нужно очистить рекомбинантную ДНК от ДНК клеток хозяина, РНК и белков. Эта процедура довольно проста и включает экстракцию, физическое разделение компонентов и ферментативное расщепление РНК и белков. Так, плазмидную ДНК можно отделить от геномной благодаря ее малому размеру и кольцевой структуре. Фаговую ДНК можно получить в свободном от клеточной ДНК виде путем очистки фаговых частиц и последующего выделения из них ДНК.
макроструктура клонированной вставки
Для определения макроструктуры вставки сначала устанавливают ее размер и положение сайтов для рестриктирующих эндонуклеаз. Поскольку клонированные фрагменты геномной ДНК часто бывают длиннее или короче, чем интересующая нас область, необходимо также определить положение и длину нужного сегмента внутри вставки.
а. Размер вставки
Размер векторной молекулы обычно бывает известен, поэтому размер вставки можно оценить, зная полную длину рекомбинантной молекулы и предположив, что никакого делетирования во время клонирования не произошло. Размер многих рекомбинантных молекул можно определить с помощью гель-электрофореза. Чтобы получить точные данные, кольцевые рекомбинантные молекулы превращают в линейные, поскольку электрофоретическая подвижность кольцевых структур зависит от степени их сверхспиральности. Стандартные электрофоретические методы непригодны для тестирования молекул, длина которых превышает 15 т.п. н, поскольку крупные молекулы мигрируют в геле слишком медленно. Поэтому размер многих рекомбинантных молекул не поддается прямому определению с помощью электрофореза, если только не используется импульсный электрофорез. Альтернативный способ оценки размера рекомбинантных молекул состоит в измерении их длины с помощью электронной микроскопии. Здесь тоже очень важны стандартные размеры, поскольку число пар оснований в сегменте определенной длины на электронной микрофотографии может зависеть от конфигурации молекулы и способа приготовления препарата.
Другой подход к определению размера вставки состоит в обращении тех реакций, которые использовались при конструировании рекомбинантных молекул, т.е. в вырезании вставок с помощью соответствующих эндонуклеаз рестрикции. Размер линейной вставки определяют с помощью гель-электрофореза. Поскольку карта сайтов для эндонуклеаз рестрикции в векторной молекуле обычно бывает известна, идентифицировать встроенный фрагмент довольно легко. Если при лигировании произошла элиминация эндонуклеазных сайтов на концах вставки, можно использовать альтернативную процедуру: разрезать векторную молекулу по сайтам, находящимся в участках, которые фланкируют вставку; в этом случае вставка будет вырезана вместе с сегментами вектора известных размеров. Иногда ситуация осложняется тем, что сама вставка содержит сайты узнавания для используемой эндонуклеазы. В таком случае сумма размеров всех фрагментов, образующихся из вставки, даст ее общую длину. Если вставка слишком велика, то ее разрезание и суммирование могут оказаться даже необходимыми для более точного определения длины.
Реклама

б. Картирование сайтов для рестриктирующих эндонуклеаз
В тех случаях, когда вектор имеет небольшие размеры и не слишком сложен, положение сайтов иногда можно определить непосредственно в самой рекомбинантной молекуле. Однако часто оказывается необходимым сначала вырезать вставку и очистить ее от векторных сегментов.
в. Субклонирование
Длинные клонированные вставки, входящие в состав Х - или космидного вектора, часто бывают весьма неудобными для манипулирования. Поэтому после построения частичной рестрикционной карты вставку можно разделить на более мелкие фрагменты путем субклонирования. В принципе субклонирование не отличается от других методов создания рекомбинантных ДНК, просто в этих случаях в качестве исходного генома используют рекомбинантный клон. Для субклонирования обычно применяют рестриктирующие эндонуклеазы, сайты узнавания которых состоят из четырех пар оснований, поскольку они способны разрезать ДНК во многих местах.
г. Определение положения интересующего нас сегмента во вставке
Для определения положения сегментов в небольших рекомбинантных геномах используются те же подходы, что и при нахождении специфических сегментов в больших геномах до клонирования. После построения рестрикционной карты вставки установить точную локализацию нужного сегмента не составляет труда. Для этого нужен только подходящий меченый зонд, аналогичный ДНК - или РНК-зонду, используемому для отбора клона в начале клонирования.
Овальбумин-это белок, состоящий из одной полипептидной цепи; он синтезируется в яйцеводе курицы после стимуляции стероидным гормоном эстрогеном и накапливается в яичном белке. У кур-несушек стимуляция гормоном происходит в естественных условиях, а у цыплят образование яйцевода и синтез овальбумина индуцируются при введении эстрогена. Гормон индуцирует транскрипцию гена овальбумина, в результате чего содержание овальбуминовой мРНК в клетке увеличивается практически от нуля до 50000 молекул через две-три недели и падает до 10 молекул на клетку после прекращения воздействия эстрогена. Яйцеводы кур-несушек представляют собой хороший источник овальбуминовой мРНК, поскольку она составляет в них около 50% суммарной мРНК. Очищенную мРНК используют затем для приготовления радиоактивного зонда, применяемого для идентификации клонированного гена овальбумина. Зондом может служить радиоактивно меченная одноцепочечная кДНК, синтезированная непосредственно на мРНК с помощью обратной транскиптазы, или клонированная дуплексная кДНК. С помощью таких зондов из популяции Х-векторов, содержащих вставки геномной ДНК курицы, полученные при ограниченном гидролизе эндонуклеазой EcoRI, были отобраны клонированные последовательности овальбуминового гена. Вставка, имела длину 6,8 т.п. н. Из нее образовались три EcoRI-фрагмента, при этом только фрагмент длиной 2,35 т.п. н. отжигался с овальбуминовым кДНК-зондом.
Реклама

Положение искомой последовательности в клонированном сегменте можно определить также с помощью электронной микроскопии. Для этого зонд и рекомбинантную ДНК смешивают, денатурируют и отжигают, а затем готовят препарат для микроскопирования. Комплементарные участки зонда и исследуемой молекулы образуют дуплексы, которые на электронных микрофотографиях выглядят как относительно толстые нити, а некомплементарные участки остаются одноцепочечными и имеют вид более тонких нитей. Гетеродуплекс, образующийся между овальбуминовым рекомбинантом формируются при условиях, благоприятных для гибридизации типа РНК'ДНК, а не ДНК'ДНК. В сегменте ДНК размером 2,35 т.п. н., который гибридизовался с мРНК, в образовании двухцепочечной структуры участвовали только около 550 п. н. Таким образом, гетеродуплексный анализ гораздо более информативен, чем определение положения кодирующих последовательностей в клоне. Он показывает, что, хотя мРНК содержит около 1800 п. н., в клонированном сегменте ДНК представлено менее трети длины кодирующих последовательностей гена овальбумина. Кроме того, 550 п. н. в клонированной ДНК, комплементарные мРНК, не образуют одну непрерывную последовательность, а распределены по четырем участкам, разделенным одноцепочечными петлями, т.е. кодирующие последовательности геномной ДНК чередуются с некодирующими. Как описано в разд.8.5, такие промежуточные последовательности, или интроны, типичны для эукариотических генов.
Тонкая структура клонированной вставки: нуклеотидная последовательность
Чтобы до конца выяснить структуру, функцию и происхождение клонированного сегмента ДНК, необходимо установить его первичную структуру - нуклеотидную последовательность. Быстрые и точные методы определения последовательностей ДНК были созданы вскоре после разработки методов, используемых в работе с рекомбинантными молекулами. В принципе сейчас можно провести секвенирование молекулы ДНК любой длины. Определение последовательности сегментов ДНК протяженностью в сотни и даже тысячи нуклеотидов представляет собой рутинную процедуру, а примерно до 1975 г. это была очень трудная задача. Для этого с помощью РНК-полимеразы сначала получали РНК-копии ДНК, а затем определяли нуклеотидную последовательность кРНК. Точность и полнота процесса копирования часто были недостаточны, а секвенирование РНК занимало много времени. Сейчас секвенируют саму ДНК, а для секвенирова-ния РНК обычно вначале получают кДНК-копии.
а. Общие принципы
Методы секвенирования ДНК можно разбить на две категории. В основе одних лежат химические реакции, в которых используются непосредственно фрагменты очищенной ДНК. Во втором случае используют ДНК-копии очищенных сегментов, полученные ферментативным путем. Эти подходы имеют и некоторое сходство. Прежде всего фрагменты ДНК обычно очищают, что легко осуществить с помощью клонирования. Далее, и в том, и в другом случае за один раз секвенируется только одна цепь ДНК. Для повышения точности лучше провести секвенирование каждой цепи дуплексной молекулы и сравнить результаты. За один раз используют только одну из цепей, помеченную радиоактивным изотопом. Определяют нуклеотидную последовательность только этой цепи, и именно о ней мы будем говорить в последующих разделах. Третья общая особенность указанных методов состоит в том, что в обоих случаях образуется набор радиоактивно меченных одиночных цепей всех возможных длин - от единицы до п, где п - полная длина секвенируемой молекулы.
В обоих подходах, химическом и ферментативном, полный набор фрагментов на самом деле бывает представлен в виде четырех отдельных наборов. В идеале каждый такой набор содержит все возможные фрагменты, берущие начало на одном конце цепи и продолжающиеся до очередного местоположения определенного нуклеотида. Так, один из наборов содержит все возможные цепи, начинающиеся в одном сайте и оканчивающиеся в тех местах, где встречается дезоксиаденозин. Второй набор содержит все возможные цепи, начинающиеся в том же сайте, а заканчивающиеся на всех встречающихся поочередно дезоксицитидиновых остатках. Третий и четвертый наборы представлены фрагментами, оканчивающимися на остатках тимидина или дезоксигуанозина.
Химический и ферментативный методы отличаются друг от друга способом получения четырех наборов фрагментов. В первом случае этот набор получают путем разрезания предварительно радиоактивно меченной цепи четырьмя разными способами, а во втором четыре радиоактивно меченных набора фрагментов получают путем копирования немеченой цепи ДНК. Последним этапом в обоих методах является разделение фрагментов, составляющих каждый из наборов, по длинам с помощью электрофореза в полиакриламидном геле в денатурирующих условиях. При этом удается разделить полинуклеотидные цепи, отличающиеся друг от друга только одним нуклеотидным остатком. Поскольку фрагменты несут радиоактивную метку, их легко выявить с помощью радиоавтографии, при этом достаточно лишь небольшого количества ДНК - порядка пикомолей или даже меньше. Все четыре набора, полученные из одного фрагмента
ДНК, подвергают одновременному электрофорезу на параллельных дорожках одной пластины геля. Сканируя каждую дорожку, расшифровывают всю последовательность. Используя специальные электрофоретические методы, можно разделить цепи размером от 1 до 300 или более нуклеотидных остатков. Молекулы, длина которых превышает несколько сотен нуклеотидов, фрагментируют и затем определяют нуклеотидную последовательность каждого фрагмента.
б. Химическое секвенирование
Получение наборов фрагментов, имеющих общие концы. Химическое секвенирование основано на специфической модификации различных пуриновых и пиримидиновых оснований. Эти модифицированные основания выщепляют затем из полимерной цепи с сохранением сахарофосфатного остова. Далее гидролизуют относительно нестабильные фосфодиэфирные связи, соседствующие с сайтом, где находилось удаленное модифицированное основание, в результате чего цепь разрывается. Все эти реакции, где в качестве примера рассмотрена модификация гуаниновых остатков. Представлены две реакции, одна из которых специфична в отношении только гуанина, а другая - только цитозина, и две реакции, специфичные в отношении либо пуринов, либо пиримидинов. Каждую из четырех реакций проводят с использованием отдельного препарата ДНК, в результате чего получают четыре набора фрагментов, необходимых для точного определения последовательности. Глубина каждой реакции строго ограничена, так что в каждой молекуле ДНК данного препарата в реакцию вступает в среднем только одно из чувствительных оснований. В результате популяция идентичных молекул ДНК превращается в ходе реакций, в которых участвуют случайные реакционно способные основания, в набор цепей, начинающихся в данном сайте на одном конце цепи и заканчивающихся на одном из реакционно способных оснований. Разрезание цепи происходит только по дезоксицитидиновым остаткам.
Каждая из четырех реакционных смесей на самом деле представлена двумя наборами цепей, получающихся в результате гидролиза препарата ДНК. Один включает все фрагменты, начинающиеся на 5'-конце исходной цепи, другой - все фрагменты, начинающиеся на 3'-конце. Нужную нам информацию о последовательности можно получить, используя любой из наборов, и выбор одного из них зависит от того, как был помечен исходный фрагмент - с 3' - или 5'-конца - перед проведением химических реакций, специфичных в отношении определенного основания. Немеченый материал нельзя визуализировать с помощью радиавтографии. Если исходным материалом является дуплексная ДНК, то помечена должна быть только одна из двух цепей, в противном случае конечные продукты будут представлены двумя разными наборами меченых цепей и данные будет трудно интерпретировать.
Введение метки в 5'-конец осуществляют с помощью полинуклеотидкиназы и у-32
Р. Если присутствуют 5'-концевые фосфомоноэфирные группы, то их необходимо предварительно удалить с помощью фосфомоноэстеразы. Одноцепочечные молекулы секвенируют сразу после введения метки. Дуплексную ДНК после осуществления киназной реакции разделяют на две комплементарные цепи с помощью денатурации и электрофореза. Меченый двухцепочечный фрагмент можно также разрезать на две части по определенному внутреннему эндонуклеазному сайту и образовавшиеся фрагменты разделить с помощью электрофореза. В этом случае секвенируемые молекулы на самом деле являются двухцепочечными, но одна из цепей не содержит метки и поэтому остается "невидимой".
Введение метки в 3'-концы дуплексных цепей целесообразно проводить в том случае, когда на 5'-концах имеются выступы. Удлинение более коротких 3'-концов катализируется либо ДНК-полимеразой I, либо обратной транскриптазой, использующими выступающий 5'-конец как матрицу; 32
Р включается в цепь в составе соответствующего а-32
Р-меченного дезоксинуклеозидтрифосфата. Мечение по З'-концу происходит в каждой из цепей двухцепочечной ДНК, а для секвенирования необходимо иметь препарат, в котором все молекулы несут метку только на одном конце. Поэтому меченые двухцепочечные фрагменты расщепляют рестриктазой и фракционируют субфрагменты с помощью гель-электрофореза или разделяют цепи ДНК. Однако, если концы дуплексного фрагмента не идентичны, часто оказывается возможным прямое получение одноцепочечного 32
Р-меченного конца. Например, если один из концов тупой, то его удлинения не происходит, и при соответствующем подборе а-32
Р-меченного дезоксинуклеозидтрифосфата можно пометить только один из концов.
в. Ферментативное секвенирование
Получение наборов фрагментов, имеющих общие концы. Для секвенирования ДНК с помощью ферментативного копирования необходимо иметь одноцепочечную ДНК, а также короткий полидезокси-нуклеотидный праймер, комплементарный небольшому участку ДНК. Одиночная цепь служит матрицей, а новая цепь наращивается, начиная с 3'-гидроксильной группы праймера путем присоединения дезоксирибонуклеотидтрифосфатов. При секвенировании проводят четыре типа реакций копирования, в результате чего образуются четыре набора цепей, синтез которых остановлен на остатках А, С, G или Т. Остановка синтеза происходит в результате присоединения дидезоксинуклеотида, у которого отсутствует 3'-гидроксильная группа, что препятствует дальнейшему удлинению цепи. Реакции проводят таким образом, чтобы вероятность остановки синтеза на любом другом остатке, кроме заданного, была очень мала.
ДНК-полимераза I может утилизировать 2',3'-дидезоксинуклео-зидтрифосфатные аналоги нормальных дезоксинуклеозидтрифосфатных субстратов. Как и при обычной полимеризации, соответствующий дидезоксинуклеотид присоединяется к 3'-гидроксильному концу праймерной цепи, однако образующийся при этом продукт не может служить праймером для дальнейшего роста цепи, и реакция останавливается.
Для проведения четырех разных реакций копирования комплекс "матрица-праймер" инкубируют в присутствии всех четырех обычных дезоксирибонуклеозидтрифосфатов и какого-то определенного дидезоксинуклеозидтрифосфата. Например, в одной реакции используют ddATP, dATP, dGTP, dCTP, dTTP, в другой - dATP, ddGTP, dGTP, dCTP, dTTP и т.д. Удлинение цепи с помощью матричного копирования происходит до тех пор, пока вместо очередного дезоксинуклеотида не присоединится дидезоксинуклеотид; в этот момент рост цепи прекращается. Поскольку терминация синтеза происходит в случайных сайтах, образуется набор цепей всех возможных длин, начиная с 5'-конца праймера до сайтов, соответствующих положению присоединенного дидезоксинуклеотида. Новосинтезированные цепи являются радиоактивно меченными, поскольку в реакции используется по меньшей мере один радиоактивно меченный дезоксинуклеозидтрифосфат. Часто им является а-32
-Р-дезоксинуклеозидтрифосфат, но это может быть также 35
8--дезоксинуклеозидтрифосфат. Иногда используется радиоактивно меченный праймер. Продукты четырех реакций подвергают одновременному электрофорезу на отдельных дорожках, как при химическом секвенировании. Последовательность считывают с радиоавтографа, который выглядит так же, как и радиоавтограф, полученный при использовании химического метода секвенирования.
Получение одноцепочечной ДНК для секвенирования с помощью дидезокситерминаторов. Молекулы ДНК обычно являются двухцепочечными; исключение составляют лишь одноцепочечные ДНК некоторых бактериофагов. Как мы уже говорили, физическое разделение цепей дуплекса не всегда осуществимо. Для разделения цепей дуплекс денатурируют нагреванием или обработкой щелочью и проводят гель-электрофорез или хроматографию. Разделение двух комплементарных цепей происходит, по-видимому, благодаря различиям их конформаций, обусловленным различиями во вторичной структуре. Если разделения не произошло, применяют другие методы получения одиночных цепей. Наиболее эффективный из них состоит в клонировании фрагмента ДНК в векторе, сконструированном на основе бактериофага М13. Любая клонируемая вставка всегда будет фланкирована одними и теми же последовательностями векторной ДНК, что позволяет использовать стандартные праймеры. Их синтезируют химическими методами, и всегда можно выбрать удобный праймер. Кроме того, не составляет труда получить отдельные клоны, каждый из которых содержит одну из двух комплементарных цепей ДНК, благодаря чему легко осуществить секвенирование обеих цепей.
Комбинируя клонирование в М13 и секвенирование с помощью дидезокситерминатора, мы можем получить эффективный способ определения последовательностей молекул ДНК, длина которых достигает нескольких тысяч пар нуклеотидов. Для секвенирования длинных дуплексных молекул их разрезают механически на фрагменты и затем проводят клонирование. Каждая образующаяся бляшка содержит определенные фрагменты или цепи. Случайно отбирая клоны, определяют последовательность вставок. Поскольку вставки образуются в результате разрыва исходной молекулы ДНК в случайных местах, некоторые фрагменты содержат перекрывающиеся последовательности. С помощью компьютера сравнивают последовательность каждого фрагмента с последовательностями других фрагментов, выявляют перекрывающиеся области и располагают отдельные фрагменты в определенном порядке. Таким способом были определены последовательности митохондриальной ДНК человека и ДНК Х-фага.
Другое применение методов секвенирования. Секвенирование с помощью дидезокситерминаторов может применяться для прямого определения последовательностей РНК или ДНК, причем даже в том случае, если в смеси помимо секвенируемой цепи присутствуют неродственные молекулы. Важным элементом при этом является праймер, комплементарный небольшому интересующему нас участку и лишь с малой вероятностью ассоциирующий с другими молекулами, присутствующими в смеси. Подобной специфичностью обычно обладает праймер длиной 20 оснований с уникальной последовательностью. При таких условиях с помощью обратной транскриптазы синтезируют кДНК. Этот метод применим только в том случае, если мы уже достаточно много знаем об интересующих нас РНК или ДНК, но круг таких систем все время расширяется.
Метод химического секвенирования может применяться для анализа специфических последовательностей ДНК, присутствующих в смеси наряду с большим числом разных молекул ДНК, в том числе даже если речь идет об одном гене в суммарной геномной ДНК млекопитающих. Прежде всего ДНК разрезают с помощью рестриктирующей эндонуклеазы, так что интересующий нас сегмент оказывается во фрагменте длиной в несколько сотен пар оснований. Затем проводят химические реакции, специфичные к определенным основаниям, в результате чего образуются четыре набора фрагментов. Такие наборы включают все возможные по длине фрагменты с интересующей нас областью, начинающиеся в сайте рестрикции, по которому была разрезана ДНК. Здесь же присутствует множество других фрагментов из остальной части генома. Фрагменты разделяют путем электрофореза в секвенирующем геле, затем переносят на нейлоновый фильтр и отжигают с 32
Р-меченным зондом, который комплементарен последовательности-мишени, находящейся вблизи одного из сайтов разрезания. Зонд ассоциирует со всеми фрагментами, образовавшимися начиная с одного конца последовательности-мишени, в результате чего на радиоавтограмме образуется типичная "секвенирующая лестница". Таким методом можно определить, например, нуклеотидную последовательность мутантных форм ранее клонированного гена. Он может использоваться также для определения характера метилирования специфической, уже клонированной области генома, поскольку гидразин реагирует с 5'-метилцитозиновыми остатками менее эффективно, чем с цитозиновыми и тиминовыми. О наличии 5'-метилцитозина в геле можно судить по отсутствию цитозинового остатка, представленного в соответствующем фрагменте ДНК, выделенном после клонирования и репликации в клетках Е. coli.
Компьютерный анализ нуклеотидных последовательностей ДНК
Простота разделения сегментов ДНК путем клонирования и их последующий анализ позволили определить нуклеотидную последовательность многих ДНК. Но хотя стремление биологов к получению точных в химическом отношении данных продолжало стимулировать эти исследования, информация оказывалась бесполезной, если не были выявлены какие-то особенности этих последовательностей. На первый взгляд длинные ряды оснований кажутся совершенно загадочными и не поддающимися расшифровке. К счастью, проблему расшифровки можно решить с помощью высокоэффективных компьютерных программ и для рутинного использования имеются программы на нескольких стандартных компьютерных языках. Возможности различных программ в значительной степени перекрываются. Их можно использовать при работе как на больших компьютерах, так и на мини - и микрокомпьютерах. Вообще говоря, эти программы пригодны для поиска как структурных, так и биологических особенностей последовательностей.
а. Хранение информации о первичной структуре
Для ввода и хранения данных о последовательностях и последующего поиска, проводимого исследователем вручную или с помощью специальной аналитической программы, используются стандартные процедуры редактирования. Большинство этих процедур предусматривают также возможность коррекции хранящейся информации. Ввод данных часто осуществляют путем типирования последовательности в компьютерном терминале после прочтения и регистрации данных секвенирующего геля. Однако имеются также программы для прямого ввода исходных данных. В принципе такие программы позволяют свести к минимуму ошибки, допускаемые при неправильном считывании гелей или неточной регистрации данных. Существует два подхода к такой автоматизации. В первом случае детектор автоматически сканирует радиоавтографы и передает данные непосредственно в компьютер. Во втором данные автоматически регистрируются от сенсора, управляемого вручную. При втором подходе исследователь является арбитром при любых затруднениях.
б. Структурный анализ
Первичная структура. Если имеются данные о нуклеотидной последовательности одной цепи, то большинство программ позволяют построить комплементарную цепь, вычислить нуклеотидный состав, выявить участки, богатые пуринами, пиримидинами или определенными сочетаниями оснований, и определить частоту встречаемости различных динуклеотидов. Могут быть выявлены специфические субпоследовательности в пределах определенного сегмента, что часто используется для нахождения сайтов для рестриктирующих эндонуклеаз. В программу введена информация о сайтах узнавания для известных ферментов, и по одной команде выдаются сведения о положении этих сайтов для каждого из ферментов, числе ожидаемых фрагментов, образующихся при расщеплении ими ДНК, размере каждого фрагмента в парах оснований и его процентном отношении к общей длине сегмента, а также о нуклеотидных остатках, соответствующих концам каждого фрагмента. Существуют также программы, которые могут предсказать, какие продукты будут получены при совместном действии двух или нескольких эндонуклеаз. При этом предсказания делаются как для линейных, так и для кольцевых молекул. Полученная информация может использоваться для подтверждения данных секвенирования путем сравнительного анализа ожидаемого и реального результатов действия эндонуклеаз.
Имеющиеся программы осуществляют также поиск характерных особенностей последовательностей, включая прямые и обратные повторы. Выявляются не только полностью совпадающие сегменты, но и сегменты с той или иной степенью несовпадения; для этого допускаются определенные отклонения параметров, заложенных в программу, от фиксированного значения. В примере перечислены все повторы длиной не менее шести пар оснований и гомологичные не менее чем на 75%. Предполагается, что максимальный размер образующихся петель равен двум основаниям.
Можно получить данные о гомологии или частичной гомологии различных последовательностей. Эти данные могут касаться структуры одного и того же гена у двух разных организмов или родственных генов одного организма. Такой сравнительный анализ широко используется при изучении эволюции на молекулярном уровне. Для сравнения двух последовательностей между собой или одной последовательности с группой других последовательностей разработаны специальные алгоритмы. Может использоваться описанный ранее вероятностный анализ. По результатам сравнения информации о последовательностях, полученной в разных лабораториях, был создан центральный банк данных, из которого всегда можно затребовать информацию о тех или иных последовательностях. К 1989 г. в банках имелась информация о последовательностях более 20 миллионов пар оснований, представляющая данные о многих генах и организмах.
Помимо этого общего применения, обнаружение сходства различных последовательностей является составной частью секвенирования очень длинных последовательностей с помощью дидезокси-метода, объединенного с клонированием в фаге М13. Используя компьютер для обнаружения перекрывающихся последовательностей, мы не только делаем работу менее утомительной, но и можем быстро решить, следует ли нам пытаться получить дополнительные данные.
Вторичная структура. Разработаны программы, позволяющие предсказать стабильную внутримолекулярную вторичную структуру одноцепочечных РНК или ДНК. Они позволяют, например, построить модель укладки цепи тРНК и рРНК, и многие из таких моделей получили экспериментальное подтверждение в опытах с использованием нуклеаз, специфичных к одноцепочечным участкам. Расчеты основаны на предположениях о вероятности образования определенных пар оснований, на термодинамических свойствах разных пар оснований и данных о стабильности спирали.
в. Биологическое значение
С помощью компьютерных программ можно перевести информацию с языка нуклеотидов на язык аминокислот в соответствии с правилами генетического кода. При этом указываются все три возможные рамки считывания и стоп-кодоны трансляции. В тех случаях, когда аминокислотная последовательность кодируемого полипептида известна, идентифицировать правильную рамку считывания не составляет труда. В противном случае правильную рамку можно выбрать, исходя из ее длины. Рамки, в которых часто встречаются стоп-кодоны, вряд ли могут считаться правильными. Показаны часть генома SV40, кодирующая участок вблизи г\ГИ2
-конца малого и большого Т-антигенов, и полученные с помощью компьютера аминокислотные последовательности для всех трех рамок считывания. Правильной является третья рамка. Можно также рассчитать частоту, с которой встречается каждый кодон, и таким образом выявить предпочтительные кодоны для определенных аминокислот. Обращение этой процедуры позволяет воссоздать возможные нуклеотидные последовательности, исходя из известной аминокислотной последовательности полипептида, хотя эта задача не имеет однозначного решения вследствие вырожденности генетического кода. Центральный банк данных содержит информацию об аминокислотных последовательностях всех проанализированных полипептидов, что позволяет сравнивать последовательности изучаемого и известных белков.
С помощью компьютеров можно вести поиск специфических нуклеотидных последовательностей, например промоторов или участков связывания с рибосомами. Они позволяют также обнаружить последовательности со специфическими свойствами. Например, наличие сходных последовательностей в разных неродственных во всех других отношениях генах или вблизи них наводит на мысль об их возможной регуляторной роли. Точно так же присутствие гомологичных последовательностей в кодирующих областях различных генов может свидетельствовать об общности эволюционного происхождения этих генов. Данный вопрос детально рассмотрен в ч. III книги. Однако важно осознавать, что статистический анализ еще не является достаточным для того, чтобы делать вывод о биологической роли той или иной последовательности. Об этом можно говорить только после проведения независимого биологического исследования.
Определение положения клонированных сегментов в геномах
В идеале установление положения клонированного сегмента ДНК означает определение фланкирующих его последовательностей и того хромосомного сайта, по которому этот сегмент был включен в хромосому. Конечная цель такого картирования состоит в полном описании структуры хромосомы.
а. Молекулярная локализация
Характеристика клонированного сегмента. Прежде всего необходимо доказать, что клонированный сегмент действительно является репликой определенного геномного сегмента. Существует ли в геноме гомологичный сегмент с точно так же расположенными сайтами для рестриктирующих эндонуклеаз? Этот вопрос имеет важное значение, поскольку во время репликации в системе хозяин-вектор иногда происходит клонирование случайных последовательностей, образовавшихся в результате рекомбинаций, делеций или мутаций. Основной подход состоит в использовании клонированной ДНК в качестве зонда для анализа продуктов эндонуклеазного расщепления суммарной геномной ДНК, из которой произошел данный клонированный сегмент. Такой подход аналогичен описанному в разд.6.1. б с тем отличием, что зондом в данном случае является клонированная вставка, меченная 32
Р. В эксперименте Б, клонированный зонд представлял собой субклонированный сегмент овальбуминового гена; материал, подвергнутый электрофорезу, - это coRI-гидролизат ДНК курицы. С зондом гибридизовался материал только одной полосы, представляющий собой фрагменты длиной 2,35 т.п. н. Этот эксперимент показывает, что 1) клонированная вставка в самом деле является фрагментом ДНК курицы;
2) в геноме гибридизующийся сегмент расположен между двумя EcoRI-сайтами, находящимися друг от друга на расстоянии 2,35 т.п. н.;
3) ни один из фрагментов любого другого размера не гибридизуется с зондом. Таким образом, клонированный фрагмент длиной 2,35 т.п. н., по-видимому, представляет собой истинную геномную последовательность и два аллеля в диплоиде являются идентичными, поскольку локализация этих EcoRI-сайтов совпадает.
Еще одна последовательность овальбуминового гена обнаружена в EcoRI-фрагменте длиной 1,8 т.п. н. После того как он был клонирован и использован в качестве зонда для анализа EcoRI-гидролизата ДНК курицы, были обнаружены три гибридизующихся фрагмента длиной 1,8; 1,3; 0,5 т.п. н. соответственно. Эти данные свидетельствуют о неидентичности двух аллельных овальбуминовых генов. Один из них содержит дополнительный EcoRI-сайт, разделяющий фрагмент длиной 1,8 т.п. н. на две части. Интересно, что столь простая процедура лежит в основе генетического анализа. Наследование двух или более аллелей легко прослеживается с помощью эндонуклеазного расщепления, электрофореза, ДНК-блоттинга и гибридизации между соответствующим зондом и ДНК, выделенной из разных источников. Единственным требованием является наличие полиморфных эндонуклеазных сайтов в аллелях любого гена или сегмента ДНК. Для проведения таких экспериментов достаточное количество ДНК можно получить из очень небольшого кусочка ткани или из нескольких миллилитров крови, поэтому метод может использоваться для анализа ДНК большинства видов, в том числе и человека.
Построение карты молекулы. Имея сравнительно короткие сегменты молекулы ДНК, такие, например, как ранее описанный субклонированный фрагмент длиной 2,35 т.п. н., можно построить детальную карту молекулы вплоть до установления ее нуклеотидной последовательности. Более длинные клонированные последовательности, подобные тем, которые присутствуют в Х-векторах или космидах, часто содержат несколько генов, а также другие сегменты ДНК, и могут использоваться для расширения карты. Так, субклонированный фрагмент овальбуминового гена длиной 2,35 т.п. н. служил зондом для поиска гомологичных последовательностей в космидной библиотеке, сконструированной из геномной ДНК курицы. Были отобраны две космиды с перекрывающимися последовательностями только в области зонда; вместе они составляли участок генома длиной 46 т.п. н., из которых 7,7 т.п. н. принадлежали самому гену овальбумина. Область, содержащая овальбуминовый ген, была идентифицирована благодаря образованию гетеродуплекса с овальбуминовой кДНК. Неожиданно обнаружилось, что два других участка сегмента длиной 46 т.п. н. также гибридизуются с овальбуминовой кДНК, хотя и в меньшей степени. При отжиге РНК из яйцевода курицы с космидами эти два участка гибридизовались также с двумя другими мРНК, отличными от овальбуминовой. Было высказано предположение, что в сегменте длиной 46 т.п. н. локализованы еще два гена, последовательность которых сходна с последовательностью гена овальбумина. Данное предположение получило подтверждение после проведения рестрикционного и гетеродуплексного анализа, а также после определения нуклеотидной последовательности. Эти два гена, обозначенные как X и Y, транскрибируются, как и овальбуминовый ген, в присутствии стероидных гормонов.
Методы, использующиеся при картировании овальбуминового гена и генов X и Y, лежат в основе широко распространенного подхода к построению детальных генетических карт, получившего название прогулки по хромосоме. Уникальный сегмент ДНК, примыкающий к одному из концов клонированного фрагмента, очищают и используют для зондирования библиотеки геномной ДНК. Одни гибридизующиеся клоны полностью перекрываются с исходным клонированным сегментом, другие же включают новые сегменты, в результате чего карта расширяется. При повторении этой процедуры совершается "прогулка" на большее расстояние. Данный метод не очень удобен для картирования тех геномов, которые содержат большое число рассеянных повторов, поскольку в этом случае трудно очистить уникальные последовательности, используемые в качестве зондов.
б. Хромосомная локализация
Классическое генетическое картирование основывается на получении определенных мутаций и анализе частот рекомбинаций. У Drosophila генетические карты удалось расширить и уточнить путем установления корреляций между генетическими данными и хромосомными аберрациями типа делеций, инверсий и транслокаций, которые визуально проявляются как изменения в характере исчерченности политенных хромосом. Однако подобные методы непригодны для анализа хромосом большинства растений и животных. Генетический анализ немногочисленных популяций с большим периодом генерации весьма затруднителен, поскольку политенность встречается редко, а хромосомы довольно многочисленны, имеют небольшие размеры и с трудом поддаются идентификации. Например, у млекопитающих установление корреляции между фенотипическими изменениями и делециями, транслокациями и инверсиями позволяет локализовать лишь ограниченное число генов в специфических хромосомах или отдельных их областях. С развитием методов получения клонированных сегментов ДНК были разработаны универсальные процедуры картирования, которые не зависят от фенотипического проявления мутаций. Удалось локализовать многие гены, в том числе и гены человека, в специфических областях хромосом.
Мы рассмотрим два наиболее распространенных метода. С помощью одного из них положение клонированного сегмента ДНК устанавливают по его способности гибридизоваться со специфическим участком в практически интактных хромосомах. Второй позволяет идентифицировать хромосому, которая несет определенную последовательность, путем гибридизации подходящего меченого зонда с гибридными клетками, содержащими различные наборы лишь из нескольких хромосом данного вида. В третьем методе, описанном во введении к ч. IV, используются клонированные фрагменты ДНК для выявления полиморфизма рестрикционных сайтов. Такой полиморфный маркер применяют затем в качестве генетического маркера для построения карт сцепления эукариотических хромосом - аналогично тому, как в классическом генетическом анализе используют фенотипические маркеры.
Картирование хромосом с помощью гибридизации. РНК-зонды или денатурированные ДНК-зонды могут гибридизоваться с соответствующими последовательностями денатурированной ДНК, входящими в состав хромосом с характерными морфологическими признаками. Положение радиоактивно меченного зонда определяют, нанеся на препарат, подготовленный для микроскопического исследования, чувствительную эмульсию; в том месте эмульсии, которое контактирует с гибридным участком, появляются темные зерна серебра. Проведя микроскопическое исследование всего препарата, можно локализовать зонд. Довольно точные данные удается получить в случае больших политенных хромосом Drosophila в основном благодаря тому, что положение темных областей можно соотнести с характером исчерченности хромосом.
Однако определение положения единичных генов в небольших и весьма многочисленных хромосомах растений и позвоночных путем гибридизации insitu затруднено по двум причинам. Во-первых, не всегда легко идентифицировать отдельные хромосомы.
Это зависит от их размера, положения центромеры и характерного рисунка из темных и светлых полос, образующегося после окрашивания фиксированных метафазных хромосом определенными красителями. Кроме того, сами полосы захватывают слишком большие отрезки ДНК. Например, 3"109
пар нулеотидов гаплоидного генома человека содержатся менее чем в 1000 различных полосах, а у растений это число даже меньше. В результате с помощью чувствительной эмульсии можно установить лишь, в какой области хромосомы находится интересующий нас сегмент. Более того, размер зерен серебра и его разброс уменьшают разрешающую способность примерно до 107
пар оснований.
Вторая проблема-чувствительность зондов. Масса одной копии фрагмента дуплексной ДНК длиной 1 т.п. н. составляет примерно 10-12
мкг. Радиоактивно меченные зонды, полученные с помощью ник-трансляции, редко имеют удельную радиоактивность, значительно превышающую 108
расп. /мин на 1 мкг. Гибридизация такого зонда с одним сопоставимым сегментом в хромосоме дает только 10-4
расп. /мин на 1 мкг, или примерно один распад в неделю. Чтобы достичь необходимой чувствительности, следует одновременно использовать несколько специальных приемов. Например, чтобы получить зонды с удельной радиоактивностью примерно 109
расп. /мин на 1 мкг, можно провести ник-трансляцию с использованием в качестве субстрата 125
1-5-йодСТР. Гибридизацию следует проводить в присутствии декстрансульфата, что увеличивает скорость процесса примерно в 100 раз, в результате чего усиливается сигнал. Число зерен также может возрасти, если одноцепочечные последовательности векторной ДНК, которые сцеплены с гибридизуемой эукариотической последовательностью в клонированном зонде, будут в свою очередь гибридизоваться с избытком молекул зонда. Для увеличения числа зерен серебра можно увеличить время экспозиции до нескольких недель. Таким способом удается определить положение единичных копий генов в специфических, но весьма обширных областях хромосом млекопитающих.
Картирование хромосом, основанное на получении гибридных соматических клеток. Для локализации клонированного сегмента в определенной хромосоме используют межвидовые соматические гибриды. Обычно такие гибридные соматические клетки содержат полный набор хромосом одного вида и только одну или небольшое число хромосом второго вида. Чтобы судить о наличии данного гена в определенной донорной хромосоме, анализируют группу разных линий гибридных клеток и устанавливают корреляцию между присутствием тестируемого гена и наличием специфической хромосомы.
Гибридные соматические клетки образуются при 1) слиянии клеток двух разных видов;
2) слиянии клеток одного вида с мини-клетками другого, содержащими одну или несколько донорных хромосом;
3) трансфекции реципиентных клеток препаратом очищенных хромосом донора. Чтобы элиминировать неслившиеся донорные и реципиентные клетки, применяют соответствующие селективные условия. Например, донорные клетки могут проявлять чувствительность к определенным лекарственным веществам, а реципиентные клетки могут быть мутантами, растущими лишь в особых условиях. В присутствии таких препаратов и в среде HAT растут только гибридные клетки, несущие в донорной хромосоме функциональный ген тимидинкиназы.
Если отобранные гибридные клетки выращивать в неселективных условиях, то донорные хромосомы в конце концов утратятся в силу более или менее случайных причин. Но клоны гибридных клеток можно получить в любой момент и идентифицировать резидентные донорные хромосомы по характеру исчерченности, выявлению ранее картированных последовательностей ДНК или ферментативных маркеров либо с использованием всех трех методов. Таким способом можно получить банки различных линий гибридных клеток, каждая из которых содержит определенный набор донорных хромосом.
Альтернативный подход состоит в сохранении селективных условий. Поддерживая условия, благоприятные для данного гена, в течение многих клеточных генераций, можно получить линии клеток только с одной стабильной донорной хромосомой. Другой способ получения гибридных клеток с одной донорной хромосомой состоит в трансфекции препаратом, обогащенным этой хромосомой.
При конструировании гибридов возникает вопрос, какой из видов будет служить донором, а какой - реципиентом. Ответ на него обычно можно найти лишь экспериментальным путем. Как правило, гибридные клетки "человек-мышь" утрачивают хромосомы человека.
После идентификации чужеродных хромосом в соматических клетках гибридов используют три подхода, для того чтобы установить, присутствует ли тестируемый ген в определенной хромосоме. Первый основан на выявлении корреляции между наличием этой хромосомы и экспрессией тестируемого гена с образованием определенного продукта. Для этого измеряют ферментативную активность или выявляют взаимодействие со специфическими антителами; при этом необходимо, чтобы используемые методы позволяли различать продукты генов донорных и реципиентных клеток. Например, два белка-продукта генов могут иметь разную электрофоретическую подвижность, как, например, в случае галактокиназ грызунов и приматов. При втором подходе используют гибридизацию между клонированным зондом и набором хромосом гибрида insitu; этот подход имеет уже описанные ограничения. Третий и наиболее общий подход связан с идентификацией искомого гена путем отжига соответствующего ДНК - или РНК-зонда с ДНК, выделенной из гибридных клеток и перенесенной на нитроцеллюлозный фильтр. Отжиг с эндонуклеазными фрагментами часто позволяет выявить донорный ген во фрагментах, имеющих размеры, типичные для донорного генома, даже в тех случаях, если гены донора и реципиента являются перекрестно гибридизующимися. Поскольку трудно получить гибриды с одной донорной хромосомой, обычно исследуют целый набор гибридных клеток, которые содержат разные донорные хромосомы. Если этот набор достаточно обширен, то присутствие последовательности зонда обычно можно связать с присутствием определенной хромосомы.
Используют также гибридные соматические клетки, содержащие лишь часть определенной хромосомы донора. Например, некоторые линии донорных клеток содержат хромосому с делецией или хромосому, несущую небольшой транслоцированный участок из другой хромосомы, и за прошедшие годы были выделены и охарактеризованы многие линии гибридных клеток, содержащие делетированные или составные донорные хромосомы. Например, с помощью таких гибридных клеток гены тяжелой цепи иммуноглобулина человека были локализованы в положении 32 длинного плеча хромосомы 14. Были проанализированы две линии гибридных клеток мыши и человека, каждая из которых содержала одну из пары реципрокных транслокаций между хромосомой 14 и Х-хромосомой. Зонд, представляющий собой клонированный ген иммуноглобулина, гибридизовался только с ДНК из гибрида, содержащего часть хромосомы 14, соответствующую q32.
В результате анализа клонов из библиотек, содержащих ДНК одной хромосомы, были получены детальные карты участков индивидуальных хромосом. Наиболее прямой путь получения таких библиотек состоит в использовании отдельных очищенных хромосом. Другой подход основан на использовании ДНК из гибридных соматических клеток, содержащих только одну донорную хромосому. Из такой библиотеки могут быть отобраны рекомбинанты, содержащие последовательности донорной ДНК, по их способности гибридизоваться с видоспецифичными зондами.
в. Нестабильность при клонировании
Как правило, клонированные вставки представляют собой точные реплики геномных последовательностей, однако иногда во время клонирования в этих последовательностях происходят изменения. Несоответствие размеров клонированной вставки и сегмента геномной ДНК чаще всего бывает обусловлено рекомбинациями, делециями или вставками, произошедшими во время клонирования. Нередко наблюдаются вариации в размерах вставки или в ее рестрикционной карте при повторном выделении. Если клонированный фрагмент содержит тандемные повторы, то при расщеплении рекомбинантной ДНК часто получается сложная смесь рестрикционных фрагментов. Например, некоторые фрагменты могут быть представлены в количестве меньшем, чем моль-эквивалент. Это тоже свидетельствует о происшедших во время репликации рекомбинантной молекулы перестройках, что приводит к образованию смешанной популяции рекомбинантов. Перестройка состоит в гомологичной рекомбинации, сопровождающейся делециями и амплификациями клонированных тандемно повторяющихся единиц. Подобная нестабильность клонированных повторов в Е. сой-системах хозяин-вектор уменьшается, но не устраняется полностью в клетках, мутантных по функциям рекомбинации.
Определение числа копий данной последовательности в геноме
Ген овальбумина присутствует в гаплоидном геноме курицы в единственном экземпляре, однако многие гены и некоторые другие сегменты ДНК встречаются в геноме множество раз. Типичными примерами геномов с многократно повторяющимися последовательностями ДНК являются эукариотические геномы. Иногда несколько копий в таком геноме бывают абсолютно идентичны, и тогда при отжиге смеси рестрикционных фрагментов геномной ДНК с меченым гомологичным зондом на электрофореграмме наблюдается одна полоса, четко видимая даже в том случае, когда количество расщепляемой ДНК не превышает 1 мкг. Обычно для обнаружения сегмента, представленного в геноме млекопитающих лишь однократно, требуется не менее 10 мкг ДНК, поэтому появление полосы при количестве ДНК 1 мкг и менее свидетельствует о том, что данный сегмент представлен в геноме многократно. Если разные копии последовательности различаются по одной или нескольким парам оснований, то у них могут быть и неодинаковые сайты для рестриктирующих эндонуклеаз. В результате на радиоавтограмме будет присутствовать множество полос или она будет представлять собой одно размытое темное пятно; это зависит от числа копий повторяющейся последовательности.
а. Оценка числа копий с помощью гибридизации ДНК-ДНК
Число копий можно оценить, проведя денситометрический анализ радиоавтограммы. При этом для повышения точности в отдельные лунки на этой же пластине геля вносят известное количество немеченой клонированной ДНК как образец геномной ДНК. Плотность радиоавтографа линейно зависит от количества ДНК в геле, поскольку зонд присутствует в избытке. Сравнивая между собой эти показатели для геномной ДНК и ДНК стандарта, мы можем оценить число копий.
б. Оценка числа копий по кинетике реассоциации ДНК
Более точную информацию о числе копий можно получить, исследуя кинетику реассоциации ДНК. Кинетические методы стали применяться в этой области по крайней мере на десять лет раньше методов, описанных в разд.7.5. а, и именно с их помощью были получены первые данные о том, что эукариотические геномы содержат много повторов. Для того чтобы понять суть кинетического метода, необходимо вывести несколько уравнений.
Скорость реассоциации отдельных комплементарных цепей в растворе зависит от концентрации ДНК и подчиняется кинетике второго порядка. Если
С0
и С-это суммарные концентрации денатурированной ДНК в момент времени t0
и в момент времени t после начала гибридизации соответственно, то скорость реакции описывается уравнением
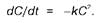
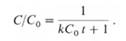
Хотя, чем больше размер генома, тем меньше истинная концентрация каждого гибридизующегося фрагмента. Другими словами, в более сложном геноме каждый гибридизующийся фрагмент составляет меньшую часть суммарной ДНК и поэтому гибридизуется при более высоких значениях Cot. Таким образом, кинетические кривые позволяют оценить размер генома, если у нас имеется препарат геномной ДНК известного размера, который может служить стандартом, и если кривые соответствуют кинетике второго порядка. Если Na
и Nb
-размеры геномов а и b, то
где С - число молей мононуклеотидов на 1 л, t - время в секундах.
Величина обычно обозначается словом, которое пишется и произносится как "Cot".
Как и во многих экспериментах по гибридизации, ДНК перед денатурацией и реассоциацией была разрезана на фрагменты длиной около 400 пар оснований. Обратите внимание на то, что шкала Cot логарифмическая; это облегчает сравнительный анализ данных. Соответствие кинетических кривых простой реакции второго порядка следует из того, что ренатурация на 90% укладывается в интервал значений Cot, охватывающий два порядка величины.
Конкретные значения Cot0.5
зависят от общего размера генома, поскольку С - это суммарная кон-
Характер кинетических кривых второго порядка для ДНК SV40, Т4 и E. coli позволяет сделать вывод о том, что каждый геномный сегмент представлен в геноме только один раз. Аналогичные кривые для эукариотической геномной ДНК имеют более сложную форму. В качестве примера приведены данные для ДНК Drosophila. Гибридизация протекает в широком диапазоне значений Cot, поскольку многие сегменты ДНК повторяются, причем одни из них многократно, другие менее часто, а третьи - лишь несколько раз. Те сегменты, которые встречаются в геноме только один раз, имеют низкую концентрацию и гибридизуются при самых высоких значениях Cot. Большинство высокоповторяющихся последовательностей, концентрация которых наиболее высока, гибридизуются очень быстро и характеризуются самыми низкими значениями Cot. Предположив, что гибридизация сегментов ДНК, представленных в единственном числе, следует кинетике второго порядка, можно из сложных Cot-кривых с помощью метода наименьших квадратов выделить отдельную кривую для однокопийной последовательности и оценить ее Cot.
Для таких последовательностей Drosophila значение Cot05
равно 28,6 моль"с/л при экспериментальных условиях, которые использовались в случае. Это значение принято за стандартное; сравнивая с ним значение Cot0.5
для какого-то клонированного фрагмента ДНК Drosophila, можно рассчитать число копий этого фрагмента по формуле.
Вторая кривая относится к гибридизации клонированного фрагмента в присутствии суммарной геномной ДНК Drosophila. Клонированный фрагмент помечен радиоизотопом, благодаря чему кинетику его гибридизации можно регистрировать отдельно. Заметим, что концентрация фрагмента достаточно мала, так что его самогибридизацией можно пренебречь. Эффективная концентрация клонированной последовательности обеспечивается соответствующими последовательностями геномной ДНК. Поэтому значение C0
t по оси абсцисс, используемое для получения значения Cot для фрагмента, рассчитывается из концентрации геномной ДНК Drosophila, а С и С0
по оси ординат относятся к фракции клонированной ДНК, которая гибридизовалась. Вот почему значения C0
t действительно относятся к геномной ДНК. Значение Cot0.5
для клонированной последовательности равно 1,06, а число копий составляет 28,6/1,06 = 27.
Для определения числа копий от одной до десяти этот метод малопригоден, поскольку для достижения высоких значений Cot требуется длительное время или очень высокие концентрации ДНК. Эта проблема еще более усложняется, если геном имеет большой размер, поскольку увеличивается значение Cot0
5
для однокопийных сегментов. Значение Cot0
5
для таких последовательностей в геноме Drosophila, имеющем размер 1,7*108
пар нуклеотидов, равно 28,6, а соответствующее значение Cot0.5
для генома млекопитающих по меньшей мере в десять раз выше.
На самом деле кинетический анализ гибридизации несколько более сложен, чем это здесь представлено. Скорость гибридизации зависит не только от концентрации ДНК, но и от температуры, ионной силы, вязкости раствора и размера гибридизующихся фрагментов. Все эти параметры должны быть строго фиксированными.
Процент гибридизовавшейся ДНК измеряют разными способами. Во-первых, можно использовать тот факт, что при определенных условиях с гидроксилапатитом связывается дуплексная, но не одноцепочечная ДНК. Во-вторых, можно определять в разные моменты времени процент ДНК, которая становится устойчивой к нуклеазе, специфичной в отношении одноцепочечной ДНК. Результаты, получаемые с помощью этих двух методов, различаются, если анализируются сложные эукариотические геномы. Нуклеазный метод позволяет определять только количество дуплексной ДНК, образовавшейся при реассоциации, а метод осаждения на гидроксилапатите дает не только количество дуплексной ДНК, но и ДНК с неспаренными одноцепочечными хвостами. Такие дуплексы образуются, в частности, в результате случайных разрывов одиночных цепей еще до реассоциации с образованием фрагментов подходящих размеров, а также благодаря наличию повторяющихся последовательностей. Нуклеаза S1 расщепляет все одиночные нереассоциировавшие цепи, а также все одноцепочечные концы дуплексов. Гидроксилапатит связывает все дуплексные цепи, включая и цепи с одноцепочечными хвостами, и поэтому дает более высокую оценку доли гибридизовавшихся цепей, чем нуклеазный метод. Иными словами, в опытах с применением гидроксилапатита мы получаем большую степень реассоциации ДНК и большую скорость реассоциации, чем в опытах с 81-нуклеазой. Эти различия уменьшаются, если используются относительно короткие фрагменты ДНК.
в. Оценка числа копий с помощью гибридизации в условиях насыщения
Еще один метод оценки числа копий состоит в определении общего количества ДНК клонированного сегмента, реассоциирующегося с известным количеством геномной ДНК. Клонированный сегмент должен присутствовать в избытке в отличие от ситуации, описанной в разд.7.5. б, где в избытке присутствовала геномная ДНК. Гибридизация в условиях насыщения особенно полезна в тех случаях, когда исследуемый сегмент присутствует в ДНК в очень малом числе копий и изучение реассоциации затруднено.
Эксперимент состоит в следующем. Возрастающее количество препарата радиоактивно меченного клонированного сегмента инкубируют с фиксированным количеством денатурированной и фрагментированной геномной ДНК и определяют количество гибридизовавшейся клонированной ДНК. Время реакции должно быть достаточным для достижения насыщения. Лучше, чтобы меченый фрагмент был одноцепочечным; это позволяет избежать его самогибридизации при относительно высоких концентрациях, необходимых для получения насыщения. В условиях насыщения все клонированные геномные сегменты связаны с зондом. Высота плато дает суммарное число гомологичных последовательностей в геноме, которое можно рассчитать исходя из удельной радиоактивности меченого фрагмента. Для повышения точности метода проводят калибровочные эксперименты, в которых к препаратам геномной ДНК добавляют известные количества немеченого клонированного фрагмента. В эксперименте количество гибридизовавшегося радиоактивного фрагмента при насыщении эквивалентно двум копиям гена на гаплоидный геном. Чтобы упростить эксперимент, препараты денатурированной геномной ДНК фиксируют в виде пятен на нитроцеллюлозном фильтре. В усовершенствованном варианте сначала определяют насыщающую концентрацию радиоактивного зонда, а затем инкубируют зонд с разным количеством геномной ДНК, фиксированной на нитроцеллюлозном фильтре.
Изменение клонированных сегментов: получение мутантов
а. Общие положения
В основе классического генетического анализа лежит получение случайных мутаций, вызывающих наследуемые фенотипические изменения. Разработка новых молекулярно-генетических методов привела к созданию так называемой обратной генетики. В охарактеризованные клонированные гены вносят специфические мутации и затем устанавливают корреляцию между изменениями в определенных участках этих генов и изменением фенотипа или локализуют регуляторные элементы. Например, если в кодирующую часть гена внести мутации, то на нем будет синтезироваться модифицированный полипептид. Это позволяет изучать влияние аминокислотной последовательности белка на его кон-формацию или ферментативную активность. Аналогично нуклеотидная замена в предполагаемом регуляторном участке может привести к подавлению или стимуляции транскрипции, что подтвердит предположение о функциональной роли данного участка.
Существуют разные способы внесения мутаций в клонированные фрагменты или небольшие геномы, но мы рассмотрим лишь немногие из них. Во всех случаях успех зависит от двух условий. Во-первых, должна быть хорошо известна структура исходной ДНК. Как минимум, необходимо знать подробную карту сайтов рестрикции, но еще лучше, если известна вся последовательность изучаемого фрагмента. Во-вторых, поскольку все методы дают некую смесь продуктов, нужный элемент следует очистить с помощью клонирования, амплифицировать и охарактеризовать. При некоторых типах мутагенеза мутации образуются в случайных местах, при других - в определенных сайтах; о последних говорят как о сайт-специфических мутациях.
б. Делеционные мутанты
Использование рестриктирующих эндонуклеаз. Внести делецию в определенный участок можно в том случае, если клонированный фрагмент ДНК содержит два близко расположенных уникальных сайта рестрикции в интересующей нас области. После эндонуклеазного расщепления фрагмента образующиеся "хвосты" отщепляют с помощью специфичной к одноцепочечным участкам нуклеазы и тупые концы вновь сшивают. Однако столь простой подход применим лишь в редких случаях. Чаще используют другой метод, при котором делецию создают в окрестности одного уникального сайта рестрикции. После эндонуклеазного разрезания фрагмента соседние нуклеотиды удаляют с помощью эндонуклеазы Bal 31 или какой-либо экзонуклеазы, например ехоШ E. coli, совместно с нуклеазой S1. При последующем лигировании образуется семейство молекул с делециями, расположенными вокруг исходного эндонуклеазного сайта. Делеционные мутанты очищают с помощью молекулярного клонирования. В одном из вариантов этого метода перед образованием кольцевой молекулы к концам присоединяют рестриктазные линкеры. При этом в молекулу вводятся эндонуклеазные сайты, которые могут оказаться полезными для исследования ее свойств, секвенирования и последующего моделирования.
При помощи более сложных манипуляций может быть получен целый набор делеций разной длины, берущих начало от одного исходного сайта. Для этого рекомбинантную молекулу обрабатывают двумя способами. Из одной ее части удаляют с помощью двух рестриктирующих эндонуклеаз большой сегмент, включающий мишень для делеций. Вторую часть линеаризуют с помощью одного из двух ферментов и затем обрабатывают ферментом Bal 31. Далее в результате разрезания вторым ферментом получают набор фрагментов уменьшающейся длины. Лигирование этих фрагментов с частью А и последующее клонирование дает набор молекул с делециями, начинающимися в сайте RE1.
Использование неспецифических эндонуклеаз. В сверхспиральные кольцевые молекулы могут быть внесены единичные разрывы с помощью неспецифических эндонуклеаз, например ДНКазы I. При этом образуется целый набор линейных молекул с разрывами в разных сайтах. После внесения разрыва и расширения делетированной области осуществляют лигирование с помощью методов, описанных ранее для устранения эндонуклеазных разрывов. При таком подходе используют некоторые интересные свойства ДНКазы I: в присутствии Мп2
+ этот фермент расщепляет сразу обе цепи дуплексной ДНК, а в присутствии Mg2
+ гидролизует за один раз только одну цепь, в результате чего в ДНК появляются одноцепочечные пробелы. Кроме того, фермент расщепляет сверхспиральную ДНК быстрее, чем линейную дуплексную, вследствие локальной неупорядоченности ДНК в сверхспиральном состоянии. Поэтому после непродолжительной обработки сверхспиральной ДНК ДНКазой I в присутствии Mg2
+ образуются в основном полноразмерные линейные дуплексные молекулы. Обработка их с помощью Bal 31 или экзонуклеазы, а затем 81-нуклеазы расширяет образовавшийся ранее пробел. После лигирования отдельные продукты можно получить с помощью молекулярного клонирования.
в. Инсерционные мутанты
Принципы конструирования инсерционных мутантов сходны с описанными выше для делеционных мутантов. Клонированный сегмент ДНК расщепляют по одному из сайтов с помощью рестриктирующей эндонуклеазы или неспецифической эндонуклеазы. При необходимости заполняют пробелы на концах образовавшейся линейной молекулы или отщепляют одноцепочечные "хвосты" с помощью нуклеазы и осуществляют лигирование в присутствии сегмента, который хотят встроить в молекулу. В качестве вставки может использоваться синтетический фрагмент, содержащий множество сайтов для рестрикционных эндонуклеаз, - так называемый полилинкер. Если первое расщепление было неспецифичным, то появление новых сайтов рестрикции в наборе клонированных мутантов поможет построить физическую карту мутаций.
г. Точечные мутации
Химический мутагенез. Для получения точечных мутаций в определенном участке молекулы чаще всего используют дуплексную кольцевую ДНК, содержащую короткий одноцепочечный участок. Один из способов создания таких сайт-специфических пробелов состоит в обработке сверхспиральной ДНК соответствующей рестриктирующей эндонуклеазой в присутствии бромистого этидия, который встраивается между плоскостями пар оснований и вносит нарушения в структуру дуплекса. При этих условиях многие рестриктирующие эндонуклеазы разрезают только одну из цепей в соответствующих сайтах. По-видимому, при встраивании бромистого этидия в обычную дуплексную молекулу ДНК разрезания вообще не происходит, а в сверхспиральной молекуле разрезается только одна цепь. Не все рестриктирующие эндонуклеазы ведут себя подобным образом, но все же число их достаточно велико. После разрезания молекулы с помощью экзонуклеазы в дуплексной молекуле создают небольшой одноцепочечный пробел в месте разреза. Альтернативный способ получения дуплексной молекулы с пробелом состоит в использовании векторной системы на основе фага М13. Для этого создают одноцепочечный М13-рекомбинант, содержащий нужный сегмент, а также другой, двухцепочечный, рекомбинант, содержащий такой же сегмент, но с делецией. Этот двухцепочечный рекомбинант денатурируют и реассоциируют с одноцепочечным, в результате чего образуется гетеродуплекс с пробелом.
Если ДНК, содержащую одноцепочечный пробел, обработать бисульфитом натрия, то в одноцепочечном участке произойдет дезаминирование остатков цитозина с образованием урацила, т.е. дезаминируются только определенные остатки цитозина. Если пробел невелик, а число дезаминированных остатков цитозина ограничено благодаря малому времени реакции и низкой концентрации бисульфита, то возникает очень небольшое число специфических мутаций. Далее пробел заполняют с помощью ДНК-полимеразы и осуществляют лигирование. Вместо исходных С"С-пар в молекуле ДНК теперь содержатся пары и"А, которые после репликации превращаются в Т"А-пары.
Мутагенное копирование. Специфические мутации другого типа можно получить, если при заполнении пробела вместо нормального дезоксирибонуклеозидтрифосфата использовать его мутагенный аналог. ДНК-полимераза I способна использовать в качестве субстратов различные аналоги обычных дезоксирибонуклеозидтрифосфатов. Некоторые из них являются мутагенами. Например,] М6
-гидро-ксидезоксицитидин-5'-трифосфат может включаться в синтезируемую цепь в положение, соответствующее А или G в матричной цепи в зависимости от того, находится ли он в иминной или аминной форме соответственно. Если пробел в дуплексной ДНК заполняется с помощью HO-dCTP вместо dTTP, то напротив А будет находиться HO-dC. После трансфекции и репликации наряду с нормальными формами будут обнаруживаться мутантные геномы с транзициями Т • А - " С • G. Проведя отбор, эти мутанты можно клонировать. Аналогично замещение dCTP на HO-dCTP приводит к транзициям С • G - > Т • А.
Сайт-специфический мутагенез с применением синтетических олигодезоксинуклеотидов. Олигодезоксинуклеотиды можно синтезировать в больших количествах, что позволяет разработать достаточно универсальные методы получения сайт-специфических точечных мутаций в клонированных сегментах ДНК. В основе одного из таких методов лежит образование гетеродуплекса между одноцепочечным синтетическим олигодезоксирибонуклеотидом, содержащим мутантную последовательность, и комплементарной одноцепочечной рекомбинантной векторной ДНК, несущей соответствующий сегмент дикого типа. Для этого ген, в котором мы хотим получить мутацию, клонируют, например, в фаге М13 и получают одноцепочечную кольцевую рекомбинантную вирусную ДНК. Затем с этой кольцевой молекулой отжигают синтетический олигодезоксирибонуклеотид длиной от 8 до 20 нуклеотидов, содержащий мутантную последовательность. Этот олигодезоксирибонуклеотид выполняет роль праймера, а оставшийся одноцепочечным участок-роль матрицы при синтезе ДНК invitro с помощью ДНК-полимеразы I. После копирования всей кольцевой молекулы начало и конец новой цепи соединяют лигазой. Образовавшаяся дуплексная молекула содержит неспаренные основания в мутантной последовательности. Интересно, что фаговое потомство, вышедшее из одной инфицированной клетки, сегрегирует как смешанная популяция фагов дикого типа и мутантных рекомбинантов, которые можно разделить при последующем клонировании.
При втором подходе, так называемом кассетном мутагенезе, участок клонированной ДНК дикого типа замещают синтетическим дуплексным олигодезоксирибонуклеотидом, содержащим мутантную последовательность. В приведенном примере клонированная вставка встроена в дуплексный вектор типа pBR322. В простейшем случае для вырезания участка, в который мы хотим внести мутацию, используют уникальные рестрикционные сайты, встречающиеся во вставке, но не в векторе. Если такие сайты отсутствуют, приходится прибегать к дополнительным ухищрениям. Дуплексный олигонуклеотид получают путем отжига двух синтетических комплементарных цепей, каждая из которых содержит соответствующую замену основания. Кроме того, эти цепи синтезируют таким образом, чтобы дуплекс, который они образуют, имел соответствующие липкие концы. В отличие от гетеродуплексного метода, при трансфекции и клонировании измененного рекомбинанта геномов дикого типа не образуется. Однако если используют смесь синтетических олигодезоксирибонуклеотидов, содержащих альтернативные основания в мутантных участках, то смесь мутантов сегрегирует после трансфекции и ее можно разделить с помощью последующего клонирования. Такой подход оказывается полезным, если необходимо получить разные мутации в одном эксперименте. Напомним, что синтез таких смешанных олигодезоксирибонуклеотидов осуществляется простым использованием на нужной стадии химического синтеза не одного мононуклеотида, а их смеси.
Изучение функций клонированных сегментов ДНК
Нередко клонирование определенных сегментов геномной ДНК, или кДНК, осуществляют с целью выяснения функций их внутриклеточных двойников.
Исходя из результатов структурного анализа клонированных последовательностей, указывающих на присутствие в них открытых рамок считывания, можно предположить, что они содержат гены. Часто удается выявить специфические промоторные последовательности и другие регуляторные элементы. Однако, чтобы подтвердить эти данные, необходимо провести прямые функциональные исследования. При этом нужно ответить на следующие вопросы:
1. Транскрибируется ли данная последовательность только в одном или нескольких типах клеток?
2. Являются ли транскриптами молекулы мРНК?
3. Влияют ли на транскрипцию изменения, происходящие в клетке?
4. Содержит ли клонированный сегмент промоторы, терминаторы или другие регуляторные сигналы, и если да, то как они работают?
5. Какова связь между структурой клонированного сегмента и структурой внутриклеточных транскриптов?
6. Могут ли транскрипты транслироваться в полипептид? Для ответа на все эти вопросы используют различные экспериментальные подходы в зависимости от того, какая именно система анализируется.
а. Характеристика внутриклеточных транскриптов, соответствующих клонированным сегментам ДНК
Говоря о любом клонированном сегменте генома, нам прежде всего необходимо ответить на вопросы, связанные с его транскрипцией invivo. Очень важными являются также данные о структурном сходстве между клонированной кДНК и внутриклеточными родственными транскриптами. В основе соответствующих исследований лежат три метода: РНК-блоттинг, анализ с использованием специфичных к одноцепочечным ДНК нуклеаз и копирование РНК, выделенной из клеток, с помощью обратной транскриптазы. Все методы включают предварительное выделение и очистку РНК из целых клеток или специфических внутриклеточных органелл, таких, как ядро или цитоплазма. Результативность всех методов зависит от способности РНК образовывать гетеродуплексы с комплементарной клонированной ДНК.
РНК-блоттинг. Блоттинг РНК аналогичен блоттингу ДНК. Он состоит в следующем. Выделенную РНК разделяют по размерам с помощью электрофореза в агарозном геле. Обычно электрофорез проводят в условиях, способствующих денатурации РНК, чтобы свести к минимуму влияние вторичной структуры молекулы на ее электрофоретическую подвижность. Щелочные условия для этой цели не подходят ввиду лабильности фосфодиэфирных связей в молекуле РНК в этих условиях. Поэтому используют такие агенты, как глиоксаль, формальдегид или мочевину. Затем РНК переносят на иммобилизованную подложку, стараясь сохранить распределение молекул РНК. Далее используют меченую ДНК в качестве зонда для выявления на фильтре соответствующих молекул РНК. Фильтр инкубируют с ДНК в условиях, благоприятствующих гибридизации. Промыв фильтр для удаления избыточной ДНК, с помощью радиоавтографии устанавливают положение зонда, а следовательно, и положение гомологичной РНК в том геле, в котором проводился электрофорез. Таким способом выявляют продукты транскрипции клонированного сегмента ДНК. Если на параллельной дорожке этого же геля одновременно провести разделение смеси РНК или молекул одноцепочечной ДНК известного размера, то можно оценить размер транскриптов. Кроме того, РНК-блоттинг позволяет оценить количество РНК, синтезированной в клетках, из которых она получена. Метод оценки аналогичен используемому при определении числа копий ДНК на нитроцеллюлозных фильтрах. Плотность полосы на рентгеновской пленке пропорциональна количеству присутствующей гомологичной РНК. Как и ранее, желательно, чтобы меченый зонд был одноцепочечным, поскольку он не должен реассоциировать с комплементарной цепью ДНК вместо РНК.
С помощью всех этих довольно простых методов можно получить обширную информацию о функциональных свойствах клонированного сегмента ДНК. Сюда относятся не только данные о способности к транскрибированию, но и оценка числа траскриптов и ее зависимость от типа клеток или внеклеточной среды. Анализируя РНК из очищенных клеточных компонентов, можно установить, где локализуются разные транскрипты - в ядре, цитоплазме или полисомах. Часто очень важным является вопрос о полиаденилировании гомологичной РНК, поскольку полиаденилирование является характерным признаком большинства эукариотических мРНК. Разделить полиаденилированную и неполиаденилированную] РНК не составляет труда, поскольку полиаденили-рованная РНК спаривается при соответствующих условиях с poly или poly. Сами полимеры обычно фиксируют на инертной твердой подложке, что упрощает отделение несвязанной poly-PHK. Фракцию poly-PHK элюируют при денатурирующих условиях. Затем РНК из каждой фракции подвергают электрофорезу, блоттингу и
тестированию на способность гибридизоваться с клонированной ДНК. Если РНК, идентифицированная с помощью клонированной ДНК, выделена из цитоплазмы и полиаденилирована, то скорее всего она представляет собой мРНК. Идентификация становится более надежной, если, кроме того, РНК связана с полисомами.
Еще одной характеристикой мРНК, гибридизовавшихся с клонированным сегментом ДНК, является их размер. Иногда РНК имеет больший размер, чем клонированный сегмент; это означает, что в клоне представлена лишь часть гена. В других случаях РНК оказывается короче клонированного сегмента, что свидетельствует о наличии в клонированной последовательности дополнительных последовательностей, не представленных в транскрипте. Это могут быть геномные последовательности, фланкирующие транскрибируемую область, или не-кодирующие последовательности, прерывающие кодирующую область и подвергающиеся сплайсингу во время созревания мРНК. Все эти взаимоотношения между клонированным сегментом ДНК и гомологичной клеточной РНК можно установить более точно, используя специфичные к одноцепочечным ДНК нуклеазы или осуществляя обратную транскрипцию.
Исследование родства между клонированным сегментом ДНК и внутриклеточной РНК с помощью ДНК-РНК-гибридизации и специфичных к одноцепочечным ДНК нуклеаз. Эксперимент включает три этапа. На первом клонированный фрагмент ДНК метят радиоактивным изотопом и денатурируют. Можно также использовать одноцепочечную ДНК. На втором этапе ДНК гибридизуют с выделенной клеточной РНК в растворе в условиях, благоприятных для образования ДНК-РНК-гибридов. Любые последовательности ДНК, присутствующие в клонированном фрагменте, но не представленные в РНК, остаются одноцепочечными. Их удаляют с помощью нуклеазы, специфичной к одноцепочечной ДНК. И наконец, гибридные молекулы денатурируют и подвергают гель-электрофорезу. Размер образующейся радиоактивной ДНК определяют с помощью радиавтографии, сравнивая положение исследуемой ДНК с положением молекул ДНК известного размера.
Этот основной эксперимент можно проводить во многих разных вариантах, каждый из которых позволяет выявить свои особенности структуры РНК в зависимости от того, какая именно нуклеаза используется, является ли РНК суммарной или очищенной цитоплазматической ро1у-мРНК. Один из таких вариантов, где в ДНК имеется один протяженный участок, комплементарный РНК, и не спарены только концы молекулы. Метод позволяет также выявить любую область, где отсутствует комплементарность между РНК и ДНК. Во многих экспериментах такого рода используется специфичная к одноцепочечной ДНК эндонуклеаза S1, поэтому метод называют картированием.
81-картирование часто используют для установления соответствия между началом молекулы РНК и специфическим сайтом в клонированном сегменте ДНК. Во многих случаях этот сайт представляет собой сайт инициации транскрипции. Точность эксперимента значительно повышается, если используется небольшой ДНК-зонд, что позволяет измерить длину защищенного сегмента ДНК с точностью до одного нуклеотида. Для получения такого зонда обычно субклонируют строго определенные участки длинного фрагмента ДНК. Размер защищенного фрагмента ДНК определяют с помощью электрофореза в таких же гелях, что и при определении нуклеотидной последовательности.
Удлинение праймера: исследование родства между внутриклеточной РНК и клонированным фрагментом ДНК с помощью обратной транскриптазы. Для проведения этого эксперимента необходим относительно короткий ДНК-зонд, который гибридизуется с участком РНК, желательно находящимся на расстоянии не более 100 пар оснований от предполагаемого 5'-конца РНК. ДНК-зонд отжигают с РНК, и он играет роль праймера при обратной транскрипции, а матрицей служит РНК. Поскольку обратная транскриптаза прекращает копирование, когда достигает 5'-конца РНК, размер продукта позволяет установить, где начинается РНК. Обычно праймер бывает радиоактивно меченным, что позволяет легко определить положение продукта в геле. Поскольку ДНК-зонд специфичен только к конкретной РНК, исследуемый препарат может представлять собой смесь разных РНК.
б. Функциональное тестирование клонированной ДНК
Клонированные сегменты ДНК можно транскрибировать и транслировать в эукариотических системах либо invitro с использованием клеточных экстрактов или очищенных ферментов, либо invivo после введения клонированного сегмента в клетку.
Системы invitro. Для приготовления бесклеточных экстрактов, содержащих РНК-полимеразы I, II и III, обычно используют разнообразные эукариотические клетки. В анализируемом сегменте должны присутствовать последовательности, ответственные за регуляцию транскрипции; необходимы также вспомогательные белки, стимулирующие транскрипцию. При фракционировании таких экстрактов получают очищенные или частично очищенные полимеразы и факторы, с помощью которых можно детально изучить механизмы транскрипции. Для осуществления трансляции мРНК с образованием соответствующих полипептидов используют другие типы клеточных экстрактов.
Системы invivo. Для изучения функций клонированного сегмента ДНК лучше всего использовать целые клетки, поскольку это позволяет следить как за процессом транскрипции, так и за процессом трансляции. Наиболее изученной биологической тест-системой являются ооциты лягушки. Ооциты - это крупные клетки объемом до 1 мкл, которые можно многократно получать от одной лягушки. ДНК инъецируют непосредственно в ядро, при этом обычно достаточно 5 нг препарата. ДНК в комплексе с гистонами ооцита упаковывается с образованием хроматина, далее осуществляются ее транскрипция и трансляция, и в течение нескольких часов можно идентифицировать продукты экспрессии генов - мРНК и полипептиды. Для проведения эксперимента часто оказывается достаточно того количества РНК и полипептидов, которое синтезируется одним ооцитом. Во многих отношениях экспрессия инъецированных клонированных генов в ооцитах протекает нормально. Исходные ядерные транскрипты генов РНК-полимераз II и III подвергаются процессингу, а зрелые мРНК и тРНК переходят в цитоплазму.
Рекомбинантные ДНК могут быть введены и в эукариотические клетки в культуре. Молекулы можно инъецировать непосредственно в отдельные клетки, однако проще вводить клонированные молекулы в большую популяцию клеток одновременно. При этом используется методика трансфекции в связи с клонированием в эукариотических клетках. По крайней мере часть молекул, введенных таким способом, достигает ядер.
Синтез полипептидов, кодируемых клонированными сегментами эукариотической ДНК
В предыдущих разделах мы уже не раз останавливались на синтезе invitro и invivo полипептидов, кодируемых вставками в рекомбинантных векторах. Скрининг по фенотипу зависит от экспрессии интересующего нас гена в клетках хозяина. Отбор нужных клонов такими методами, как HART и гибридизационная селекция, осуществляется с помощью трансляции invitro, а функциональный анализ клонированного сегмента после введения его в исходную клетку основывается на изучении как транскрипции, так и трансляции. Цель многих экспериментов по клонированию состоит в синтезе нужного полипептида, а иногда - в получении его в количествах, достаточных для проведения фенотипического анализа. В ряде случаев цель клонирования состоит в получении антител. В этом разделе мы опишем некоторые системы хозяин-вектор, специально созданные для продукции нужного белка.
Синтез белков, предназначенных для проведения тех или иных исследований или для применения в медицине и промышленности, - это одна из самых первых задач, которые ставились при разработке технологии рекомбинантных ДНК. В настоящее время ряд поступающих в продажу ферментов, используемых для конструирования рекомбинантных молекул ДНК, в свою очередь синтезируются под контролем генов, клонированных в плазмидных векторах E. coli. Использующиеся при этом препараты имеют высокую степень очистки, относительно недороги и включают ДНК-полимеразу I, ДНК-лигазу E. coli, обратную транскриптазу. Другие белки и их мутантные формы, полученные после сайт-специфического мутагенеза соответствующих клонированных генов, используют для установления связи между структурой белка и его функцией. К ним относятся алкогольдегидрогеназа, в-лактамаза, цитохром с - и тирозил-тРНК-синтетазы. С помощью методов клонирования были синтезированы важные в клиническом отношении белки, которые трудно было получить иным способом. Сюда относятся интерфероны, инсулин и гормон роста человека. Были синтезированы белки, кодируемые некоторыми вирусами животных и простейшими. Они использовались в качестве антигенов в некоторых вакцинах.
а. Выбор системы экспрессии
Для экспрессии уже клонированного гена выбор хозяина имеет такое же важное значение, как и при самом клонировании. Наиболее подходящими хозяевами для получения белка часто оказываются клетки E. coli, поскольку с ними просто манипулировать и их легко выращивать в больших объемах. После открытия интронов, прерывающих кодирующие участки многих эукариотических генов, для использования в экспрессирующих векторах
чаще стали применять клонированные кДНК, а не сами гены. Однако при синтезе активных форм определенных эукариотических белков в клетках E. coli возникает ряд проблем. Так, первичные продукты трансляции некоторых эукариотических генов должны подвергнуться специфическим посттрансляционным модификациям, прежде чем образуются функциональные молекулы. Обычно эти модификации состоят в протеолизе, фосфорилировании, ацетилировании и гликозилировании. Ферменты, катализирующие эти реакции в клетках Е. coli, обычно отсутствуют. Отчасти для решения этой проблемы были разработаны эффективные эукариотические системы экспрессии. В животных системах хозяин-вектор в качестве кодирующих последовательностей не обязательно использовать кДНК, поскольку эти клетки способны удалять интроны.
Векторы, предназначенные для эффективной транскрипции и трансляции клонированного сегмента, содержат элементы, повышающие эффективность репликации вектора и экспрессии соответствующих генов. Репликация как таковая для экспрессии генов не требуется, но, обеспечивая образование большого числа матриц для транскрипции, она повышает содержание мРНК и полипептидов в клетке. Таким образом, экспрессирующие векторы обычно содержат точку начала репликации и кодируют некоторые другие функции, необходимые для эффективной внутриклеточной репликации и не обеспечивающиеся хозяйскими клетками. Транскрипция зависит от наличия в векторе промотора, который соответствует присутствующей в клетках РНК-полимеразе, т.е. промотора E. coli для экспрессии в клетках бактерий и промотора, используемого РНК-полимеразой II, для экспрессии в эукариотических клетках. Этот промотор должен быть расположен в векторе таким образом, чтобы клонированный ген мог встроиться в правильной ориентации и за промотором по ходу транскрипции. Эукариотические экспрессирующие векторы должны также иметь сайт для расщепления и полиаденилирования транскрипта-предшественника функциональной мРНК. Для точной трансляции в начале кодирующей области должен находиться кодон ATG, а в конце - один из стоп-кодонов. Последний обычно содержится в клонированном сегменте, но стартовый кодон часто отсутствует во вставке кДНК, поэтому его функции должен выполнять вектор. Для трансляции в Е. coli на 5'-конце на расстоянии примерно 10 нуклеотидов от инициирующего кодона должен находиться сайт связывания с рибосомой, последовательность Шайна-Дальгарно.
В экспрессирующие векторы часто бывают включены дополнительные элементы. Обычно они предназначаются для увеличения выхода полипептидов или для упрощения процедуры очистки нужного белка от других клеточных белков. Одним из основных факторов, ответственных за уменьшение выхода, является нестабильность многих белков в чужеродной клеточной среде.
Чтобы добиться экспрессии гена, обычно каждый раз приходится решать уникальную экспериментальную задачу. Поэтому многие экс-прессирующие векторы, хотя и обладают некоторыми общими свойствами, имеют свои особенности. Ниже мы рассмотрим системы, в которых используются оригинальные экспрессирующие векторы, предназначенные для конкретных целей.
б. Экспрессирующие векторы, используемые в E. coli
Векторы, сконструированные для осуществления эффективного белкового синтеза в E. coli, обычно происходят от pBR322 или другой высококопийной плазмиды; это необходимо для получения максимального числа матриц для транскрипции.
Промоторы. Большинство векторов, предназначенных для изучения экспрессии генов, содержат один из сильных промоторов, описанных в разд.3.11: Лас-промотор с соответствующим оператором, trp-промотор и оператор, а также Рь
-промотор бактериофага X. Обычно используют мутантный промотор, называемый UV5, поскольку это позволяет осуществлять транскрипцию с большей скоростью, чем в случае промотора дикого типа, а кроме того, активность сохраняется даже в отсутствие положительного эффектора САР-сАМР. Другой часто используемый промотор, lac, представляет собой синтетическую конструкцию: его - 35-последовательность представляет собой - 35-последовательность trp-промотора, а блок Прибнова - соответствующую последовательность промотора UV5. Таким образом, 1ас-промотор идентичен оптимальному промотору E. coli, структура которого была определена исходя из данных, полученных при сравнении последовательностей многих природных промоторов. Как и ожидалось, он оказался весьма эффективным.
Помимо такого достоинства, как эффективность, эти четыре промотора обладают еще одним ценным свойством: они позволяют осуществлять временной контроль инициации транскрипции, поскольку связаны с регуляторными элементами. Это очень важно, так как накопление больших количеств чужеродного для E. coli белка может подавлять рост клеток и тем самым ограничивать конечный выход белка. Впрочем, если клетки удается вырастить до высокой плотности, а затем индуцировать к оптимальной экспрессии клонированного гена, то часто можно достигнуть оптимального выхода белка. Если белок, который предполагается синтезировать, нестабилен в клетках Е. coli, то молекулы, синтезированные в начале цикла роста, вероятно, деградируют ко времени достижения культурой высокой плотности. Поэтому имеет смысл отсрочить экспрессию до того момента, когда плотность культуры повысится достаточно сильно. Если используются векторы, в которых экспрессия клонированного гена зависит от этих промоторов, то количество нужного белка обычно составляет от 1 до 10% уровня суммарного белка в клеточном экстракте.
Экспрессирующий вектор с lac-промотором. Плазмидные векторы, содержащие промотор lacUV5, часто содержат также последовательность Шайна-Дальгарно ас-оперона и кодирующие последовательности для в-галактозидазы. Поскольку экспрессия, инициируемая на участке с ас-оператором-промотором, находится под контролем ас-оперона, транскрипцию можно инициировать с помощью индуктора изопропил-в-В-тиогалактозида. В приведенном примере вместо восьмого кодона в-галактозидазного гена встроен полилинкер, что нарушает кодирующую рамку. Клетки E. coli, несущие такой вектор, не способны синтезировать активную в-галактозидазу. Две части рамки считывания можно совместить, если в один из рестриктазных сайтов полилинкера встроить фрагмент с определенной рамкой считывания. В этом случае трансляция мРНК может инициироваться в начале кодирующего участка в-галактозидазного гена, проходить через вставку и остальную часть кодирующего участка этого гена и заканчиваться на обычном стоп-кодоне. В результате клетки, содержащие плазмиду с такой вставкой, будут продуцировать активную в-галактозидазу, поскольку первые восемь аминокислот молекулы несущественны для активности фермента и чужеродные аминокислоты не будут влиять на нее. Гибридный полипептид можно очистить, при этом показателем степени очистки будет служить удельная активность в-галактозидазы. В некоторых случаях гибридный белок можно расщепить и удалить протяженную карбоксильную в-галактозидазную часть. Однако и сами гибридные белки находят применение: с их помощью получают антитела к чужеродному белку.
Экспрессирующий вектор с trp-промотором. В векторе, перед полилинкером находятся trp-промотор, оператор, последовательность Шайна-Дальгарно, аттенуатор, а также примерно 300 кодонов гена trpE. Транскрипцию можно контролировать, поместив содержащие плазмиду клетки Е. coli, выращенные до высокой плотности, в среду без триптофана и добавив затем 3-в-индолилакриловую кислоту. Последняя конкурирует с оставшимся триптофаном за связывание с trp-репрессором, однако образует неактивный комплекс, в результате чего происходит дерепрессия промотора. Как и в случае с вектором, содержащим Лас-промотор, при встраивании чужеродного гена в рамку считывания по одному из сайтов рестрикции в полилинкере синтезируется гибридный белок. Трансляция останавливается, дойдя до случайного стоп-кодона вектора.
В случае вставка кодирует обратную транскриптазу ретровируса. После трансфекции и дерепрессии в клетках E. coli синтезируется ферментативно активный гибридный белок. Для повышения стабильности обратной транскриптазы большую часть избыточных последовательностей гена trpE делетируют с помощью некоторых манипуляций и переклонирования. Несколько аминокислот trpE, остающихся на N-конце образовавшегося полипептида, слабо влияют на активность обратной транскриптазы.
Экспрессирующий вектор, использующий PL
-npo-мотор бактериофага X. Вектор позволяет синтезировать эукариотические белки, не сливающиеся с чужеродным полипептидом. Транскрипция с этого промотора подавляется X-репрессором, который образуется профагом, присутствующим в клетке-хозяине. Применение клеток, лизогенизированных фагом, который кодирует чувствительный к нагреванию репрессор, позволяет индуцировать экспрессию гена путем переноса клеток, выращенных при 30°С, в среду с температурой 42°С. Кроме промотора вектор содержит часть последовательности Шайна-Дальгарно гена cII фага X. Между промотором и этой последовательностью находится группа регуляторных элементов фаговой ДНК, которые функционируют в цис-положении, ослабляя транскрипционную полярность. Эти последовательности с антиполярным эффектом функционируют при наличии продукта гена N фага X, поставляемого лизогенизированными хозяйскими клетками, которые обычно используются с вектором этого типа. В таком векторе сегмент, содержащий связывающийся с рибосомой участок Ш-Д-последовательности, включает также кодон ATGcII-гена фага X. Более того, ATG перекрывает BamHI-сайт, унаследованный от плазмиды pBR322. Эукариотические кодирующие участки встраивают в BamHI-сайт разными способами, зависящими от кодирующей последовательности. Очень важным моментом является встраивание кодирующей последовательности таким образом, чтобы она находилась в рамке с ATG. Таким образом, вектор поставляет все регуляторные элементы, необходимые для транскрипции и трансляции, за исключением стоп-кодона, который обычно находится в пределах клонированных эукариотических кДНК.
в. Экспрессирующие векторы, используемые в клетках дрожжей
Наиболее эффективные в отношении экспрессии генов дрожжевые векторы сконструированы на основе 2 мкм-кольцевой плазмиды дрожжей. Эта плазмида обеспечивает образование в дрожжевых клетках большого числа копий рекомбинантной ДНК до тех пор, пока сегмент REP3 находится в цис-положении, а функциональные гены REP1 и REP2 - либо в цис-, либо в транс-положении. Применяются несколько разных регулируемых дрожжевых промоторов; в примере это промотор гена CYC1, кодирующего изо-1-цитохром с. Этот промотор и связанные с ним регуляторные сигналы составляют область размером в несколько сотен пар оснований, что типично для подобных сложных эукариотических областей, в которых транскрипция регулируется с помощью РНК-полимеразы II. Сегменты ДНК, трансляцию которых мы хотим осуществить, встраивают в рамку считывания в сайте. В этом случае они непосредственно прилегают к инициирующему кодону ATG гена CYC1. Сайт полиаденилирования в векторе отсутствует, но обычно кДНК содержат такой сайт, который расположен за стоп-кодоном трансляции.
г. Экспрессирующие векторы, используемые в клетках животных
Для экспрессии клонированных генов в клетках животных были сконструированы два типа векторов; в основе одного из них лежит геном SV40, а другого - геном папилломавируса крупного рогатого скота. Основные свойства этих двух систем вирусных векторов описаны в разд. 5.7. Векторы на основе папилломавирусов особенно полезны для синтеза больших количеств белка, а векторы на основе SV40 используются во многих экспериментах.
Типичные векторы содержат либо весь геном BPV, либо его часть, необходимую для стабильной трансформации хозяйских клеток, например мышиных клеток в культуре. Такие векторы часто включают эукариотический ген вместе с промотором, сигналом полиаденилирования и другими регуляторными областями. Между сайтом инициации транскрипции и стартовым кодоном трансляции находится удобный рестриктазный сайт. Когда в этот сайт встраивается определенная кодирующая последовательность, она транскрибируется с образованием соответствующей мРНК. Синтез этой мРНК начинается от стартового кодона транскрипции и заканчивается на сигнале полиаденилирования, находящемся во вставке или в векторной последовательности. Кодирующая последовательность должна содержать свой собственный стартовый и стоп-кодоны. Одним из преимуществ экспрессирующих векторов, созданных на основе BPV, является то, что трансформированные ими клетки сохраняются в культуре в течение многих месяцев. В результате непрерывного деления клеток постоянно синтезируется нужный белок. В одном из экспериментов в BPV-вектор была встроена предварительно клонированная кодирующая часть гена поверхностного антигена вируса гепатита В. Трансформированные клетки секретировали около 10 мг антигена на 1 л культуры за 24 ч.
Ферментативная амплификация сегментов ДНК и РНК
На заре развития методов молекулярного клонирования применение альтернативных способов амплификации специфических сегментов ДНК или РНК, присутствующих в больших популяциях молекул, казалось маловероятным. Однако в настоящее время такой метод разработан и широко используется. Он получил название полимеразной цепной реакции. С его помощью можно получать микрограммы ДНК-копии сегментов ДНК или РНК, даже когда они присутствуют в препарате в виде единственной молекулы.
Вся полимеразная цепная реакция осуществляется invitro с использованием ДНК-полимеразы и олигонуклеотидных праймеров, комплементарных двум 3'-концам участков, ограничивающих амплифицируемый дуплексный сегмент. Таким образом, разработка ПЦР-метода стала возможной лишь после того, как были созданы методы быстрого и недорогого химического синтеза олигонуклеотидов. Для осуществления реакции необходимо знать нуклеотидную последовательность того участка, который мы хотим амплифицировать, чтобы можно было синтезировать соответствующие олигонуклеотидные праймеры.
Полимеразная цепная реакция. Для того чтобы амплифицировать какой-то сегмент ДНК, синтезируют два олигонуклеотидных праймера, каждый из которых комплементарен одному из двух 3'-концов участков, ограничивающих амплифицируемый дуплексный сегмент. В ходе ПЦР-реакции необходимо копировать последовательность каждой из цепей, находящуюся между участками, с которыми спариваются олигонуклеотидные праймеры. После спаривания праймеров с денатурированной ДНК, содержащей амплифицируемый сегмент, осуществляют встречное удлинение праймеров с помощью ДНК-полимеразы в присутствии четырех дезоксинуклеозидтрифосфатов. Образующиеся дуплексные ДНК денатурируют, вновь спаривают с праймерами и проводят ДНК-полимеразную реакцию. Этот цикл может быть повторен примерно 60 раз с удвоением в каждом цикле количества дуплексного сегмента ДНК. За п циклов образуется 2" копий дуплексного сегмента, ограниченного праймерами.
Используя термостабильную ДНК-полимеразу, выделенную из термофильных бактерий Thermusaquaticus, можно осуществлять множество ПЦР-циклов при однократном введении фермента. Сначала смешивают ДНК, избыточное количество праймерных молекул, дезоксинуклеозидтрифосфаты и полимеразу. Цикл запускают, нагрев смесь до температуры, обеспечивающей денатурацию ДНК; затем охлаждают раствор до температуры, необходимой для отжига праймеров. Далее устанавливают температуру, при которой может происходить синтез ДНК. Весь процесс, который длится несколько часов, можно автоматизировать, используя нагреватели, работа которых регулируется с помощью компьютера.
Использование ДНК-полимеразы Т. aquaticus не только позволяет автоматизировать процесс, но дает и другое преимущество. Этот фермент наиболее активен в температурном интервале 70-75°С. При таких условиях спаривание олигонуклеотидных праймеров и ДНК происходит более специфично, чем при 37°С - оптимальный температуре для ДНК-полимеразы Е. coli. В результате ассоциация праймеров происходит более точно, что сводит к минимуму амплификацию нежелательных сегментов ДНК, особенно в присутствии всей геномной ДНК. Точность отжига повышается также при подборе оптимального температурного режима, ионной силы, длины праймера и его нуклеотидного состава. Специфичность может быть достаточно высокой, чтобы осуществлялась амплификация двух разных сегментов в одном и том же препарате ДНК в присутствии двух пар праймеров.
РНК можно амплифицировать с образованием дуплексной ДНК аналогичным образом с той лишь разницей, что в начале первого цикла необходимо синтезировать кДНК-копию РНК. Если РНК представлена молекулами мРНК, то роль одного из праймеров в ПЦР, а также в реакциях с участием обратной транскриптазы могут выполнять короткие рогу-цепочки.
Разные варианты полимеразных цепных реакций. Как мы уже говорили, для проведения ПЦР необходимо знать нуклеотидные последовательности, фланкирующие амплифицируемый сегмент. Это подразумевает, что ПЦР-метод может применяться только при наличии предварительно клонированных и секвенированных сегментов ДНК. Однако с помощью относительно простых модификаций можно значительно расширить возможности метода ПЦР. В одном из вариантов можно выделить определенный ген, если известна аминокислотная последовательность лишь короткого участка соответствующего очищенного белка. Например, синтезировав праймеры длиной 20 пар нуклеотидов на основании данных о последовательности двух концов пептидного сегмента длиной в 20 аминокислот, можно амплифицировать геномный фрагмент длиной 60 п. н. Вследствие вырожденности генетического кода при этом используют смесь праймеров с альтернативными основаниями в нужных положениях. После проведения ПЦР амплифицированный сегмент из 60 п. н. очищают с помощью гель-электрофореза или клонирования и используют в качестве зонда для идентификации интересующей нас последовательности при анализе библиотек геномной ДНК или кДНК.
Еще один пример универсальности метода ПЦР дает использование его для амплификации полной кДНК-копии мРНК исходя из данных последовательности одного небольшого участка либо соответствующего пептида, либо мРНК. Метод состоит в следующем. Суммарную мРНК копируют с образованием одноцепочечной кДНК, используя короткий рогу^Т) - праймер. Затем с помощью терминальной трансферазы к 3'-концу кДНК присоединяют "хвост"; разд.4.8); используя короткий рогу^С) - праймер, синтезируют вторую цепь ДНК. До этого момента никакой специфичности в отношении определенной мРНК не проявляется. На следующем этапе используют олигонуклеотидный праймер с такой же последовательностью, как и у короткого участка вблизи 3'-конца нужной нам мРНК. С помощью этого праймера и poly проводят ПЦР; специфичность оказывается достаточно высокой для получения практически чистого продукта, представляющего полноразмерную мРНК.
В другом варианте в 5'-концы праймеров включают последовательности, соответствующие какому-либо рестриктазному сайту. При отжиге с исходной матрицей они остаются неспаренными, но амплифицируемый фрагмент приобретает концевые рестриктазные сайты. Они могут оказаться полезными при последующем клонировании или включении в вектор для изучения экспрессии.
Применение ПЦР. Возможности метода ПЦР во всей полноте еще не раскрыты, но с его помощью уже удалось получить новые данные о структуре разных РНК, генов и геномов, а также о процессах, протекающих на геномном уровне. Одно из его очень важных применений состоит в определении характера мутаций. После того как ген клонирован, с помощью двух праймеров амплифицируют соответствующий геномный сегмент, полученный от разных особей данного вида. Секвенирование этого сегмента позволяет установить природу мутации, произошедшей в данном гене, или выявить полиморфизм, будь то замена одной пары оснований или делеция, инсерция или перестройка. Благодаря простоте метода с его помощью можно диагностировать генетические заболевания и проводить популяционно-генетические исследования. Используя праймеры, которые специфически ассоциируются с определенными мутантными формами гена, можно также классифицировать мутации.
С помощью зондов, комплементарных геномам таких инфекционных агентов, как вирусы и бактерии, могут быть идентифицированы геномы этих агентов на фоне значительного избытка ДНК клетки-хозяина. В частности, вполне реальной представляется диагностика СПИДа с помощью этого метода. Еще одно его применение состоит в анализе структуры продуктов реакций рекомбинации.
Одним из интересных и потенциально наиболее эффективных применений ПЦР является амплификация сегментов ДНК внутри отдельных клеток. Такие опыты проводились как на диплоидных клетках, так и на сперматозоидах человека. Использование сперматозоидов имеет особое значение для генетического анализа видов, включая человека, которые не могут подвергаться экспериментальному скрещиванию. Этот метод позволяет оценить частоту мейотической рекомбинации непосредственно в больших популяциях сперматозоидов, если использовать пару праймеров, специфичных в отношении полиморфных или мутантных форм сцепленных генов.
|
