Процесс растворения сопровождается разрушением структуры полимера, растворителя и возникновением новой структуры раствора [1]. В последнее время этому вопросу уделяется большое внимание. Структуру концентрированных растворов полимеров изучали в работах [2—4]. В данной работе при исследовании свойств разбавленных растворов полиарилатов рассматривали два фактора: конформации макромолекул в растворе и ориентациоиный порядок сольватирующих их молекул растворителя, о котором судили по термодинамическим параметрам раствора.
На конформацию макромолекул в растворе большое влияние оказывает равновесная жесткость цепи полимера. В данном исследовании попытались проследить, каким образом изменение строения только кислотной компоненты элементарного звена макромолекулы полиарилата скажется на равновесной жесткости его цепи, а следовательно, и на конформации макромолекулы в растворе и на структуре самого раствора. Исследовали полиарилаты следующего строения:
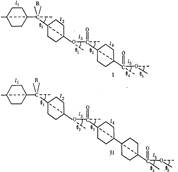
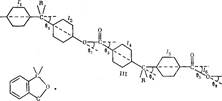
Полиарилаты синтезировали методом высокотемпературной поликонденсации е-среде высококипящего растворителя (а-хлорнафталина) при 220° и продолжительности синтеза 12 ч по методике работы [5]. Фракционирование полимеров на 12-15 фракций проводили методом распределения между двумя несмешивающимися жидкостями, в качестве растворителя использовали смесь тетрахлорэтан (ТХЭ) : фенол в соотношении 3 :1 по весу, осадитель — и-гептан.
ММ фракций и вторые вириальные коэффициенты А2
растворов измеряли методом светорассеяния на фотогониодиффузометре «Fica». Растворы очищали фильтрованием через систему фильтров 3 и 4. Перед измерением интенснвностей светорассеяния кюветы с растворами термостатировали в течение 1 ч в термостате при температуре измерения. Точность термостатирования ±0,1°. Инкремент показателя преломления растворов определяли на рефрактометре типа «Пульфриха», снабженном дифференциальной кюветой.
Растворители очищали по известным методикам [6], чистоту растворителей контролировали по показателю преломления.
Ранее нами было найдено [7], что для полимера I 6-растворителем служит ТГФ (9=22°). Предварительные опыты по температурному осаждению полимеров из раствора показали, что ТГФ можно использовать в качестве О-растворнтеля и для полимера II. Оказалось, что при повышении температуры >20° растворы становились мутными, поэтому 9-температуру искали в температурном диапазоне <20°. Полимер III в ТГФ не растворился. При комнатной температуре этот полимер хорошо растворился в ДХ, а при повышении температуры >45° полимер выпадал в осадок, что свидетельствовало о наличии НКТС. 0-температуры для всех полимеров находили экстраполяцией температурных зависимостей Л2
к 0, используя растворы нефракционированных образцов и фракций.
Реклама

Температурные зависимости удельных парциальных объемов полимеров в 6-рас-творителях определяли по методике работы [8].
Характеристические вязкости растворов исходных полимеров и их фракций определяли при помощи капиллярного вискозиметра с «висячим» уровнем в ТХЭ при 25±0,1° и в 8-условиях.
Характеристики исследованных образцов и результаты определения 9-условий даны в табл. 1.
Анализ данных по изменению А2
с температурой (рис. 1) свидетельствует о том, что для всех исследованных полимеров характерно ухудшение растворимости с повышением температуры, что подтверждает наличие НКТС. Как известно, НКТС наблюдается в системах с сильными меж-молекулярными взаимодействиями, которые могут быть обусловлены различными причинами, в частности возникновением в растворах донорно-акцепторных связей между макромолекулами полимера и молекулами
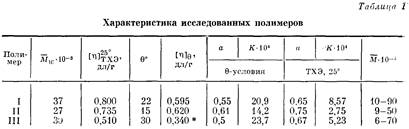
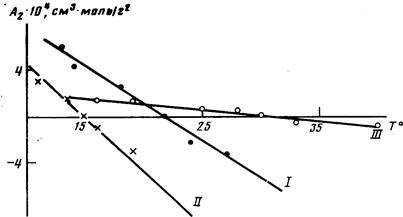
Рис. 1. Температурные зависимости Аг
для полиарилатов I—III растворителя [1].
При растворении полимеров I и II в ТГФ возможны такого рода взаимодействия между положительно заряженным атомом углерода карбонильной группы и неподеленными электронными парами атома кислорода в ТГФ. Растворение полиарилата III в ДХ может быть обусловлено донорно-акцепторными взаимодействиями между л-электронами бензольных колец полимера и свободными Зс орбиталями атома хлора растворителя. Полимеры I и II в ДХ также растворимы, однако с повышением температуры их растворимость улучшалась, о чем свидетельствовали данные по температурным зависимостям А2
: для полимера I значения А2
изменялись от —2-Ю-4
см3
моль/г2
до 1-10-4
см3
-моль/г2
в температурном диапазоне 20—35°, для полимера II от —10-10~4
см3
-•моль/г2
до 2-10-4
см3
-моль/г2
в интервале температур 25—55°.
Таким образом, полученные результаты показали, что изменение строения элементарного звена полимера влияет на процесс растворения, а следовательно, должно отразиться и на термодинамических параметрах растворов. Воспользовавшись температурной зависимостью второго ви-риального коэффициента А2
, оценили энтропийный и энтальпийный вклады в энергию взаимодействия полимер — растворитель. Согласно Флори [9], при температурах, близких к 6, справедлива следующая зависимость:
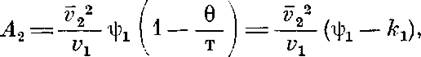
где и kiсоответствуют энтропийному и энтальпийному вкладу в энергию взаимодействия полимер — растворитель; v2
— удельный парциальный объем полимера в растворе; vi
— мольный объем растворителя.
Реклама

На рис. 2 представлены температурные зависимости параметров гр±, А-ч и разности г|)1—к,. Полученные данные показали, что процесс растворения всех трех полимеров протекает экзотермически (Ач<0) и сопровождается возникновением ориентации молекул растворителя около молекул полимера (i|)i<0). Нужно учесть, что донорно-акцепторные связи указанного выше типа не относятся к числу слабых. В области, близкой к 6-темпера-туре, наблюдается разброс экспериментальных значений и kt
как для нефракционированных образцов I и II (рис. 2), так и для фракций. Для всех трех систем характерно уменьшение разности к С) с ростом температуры, что отражает ухудшение термодинамического качества растворителя при нагревании. На рис. 2, а, б обращает на себя внимание следующий факт: в области температур ниже 8 значения ifi и kiдля всех систем остаются практически постоянными, причем для полимера III это постоянство сохраняется во всей исследованной температурной области.
Малые по величине значения параметров if>iи kiдля полимера III, по-видимому, связаны с тем, что ДХ образует слабые комплексы с полимером, поскольку его донорное число DNsbcuравно 0,1, в отличие от ТГФ,
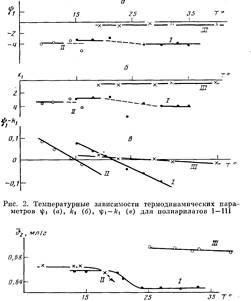
Рис. 3. Температурные зависимости Ъ2
полиарилатов I —III в растворах ТГФ (I, II) и ДХ ЦШ)
у которого ZWS
bci5=20 [10]. Это подтверждается данными по температурному изменению удельных парциальных объемов полимеров в растворах (рис. 3). Значения v2
для систем в ТГФ при температурах ниже 9 близки н остаются практически постоянными, в области 8-температуры происходит заметное уменьшение значений v2
. Для полимера III в ДХ значения v2
значительно выше, и заметных изменений во всем температурном диапазоне точно так же, как для энтропийного и энтальпийного параметров, не наблюдается. Таким образом, результаты измерения температурной зависимости v2
показали, что в растворителе, который образует слабые донорно-акцепторные связи с полимером, макромолекулярный клубок имеет более рыхлую упаковку.
Следующей задачей нашего исследования было выяснение влияния изменения строения элементарного звена полимера на конформации макромолекул в растворе. Вначале методом машинного эксперимента на ЭВМ проведено моделирование макромолекулярного клубка методом Монте-Карло по программе, описанной в работе [11]. Структурные единицы полиарилатов моделированы на основании литературных данных о строении простейших молекул, близких по составу и строению к мономерным звеньям [12]. В табл. 2 приведены длины виртуальных связей углы между ними и указана возможность свободного вращения вокруг виртуальных связей, 8-угол дополнительный к углу между виртуальными связями. Вращение вокруг связи С—О запрещено, потому что эта связь считается полу торной, так как ее длина меньше суммы ковалентных радиусов углерода и кислорода.

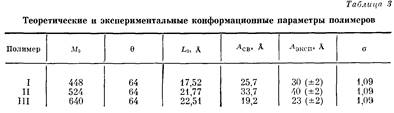
Полученные в результате машинного эксперимента конформационные параметры приведены в табл. 3.
Анализ этих данных позволяет сделать следующие выводы относительно влияния химического строения звена на конформационные параметры при свободном вращении: увеличение угла между виртуальными связями, а также возрастание длины виртуальной связи приводит к увеличению равновесной жесткости цепи, напротив замена одной виртуальной связи hна две (в полимере III) уменьшает жесткость.
Ранее нами было показано [7], что моделью для описания поведения макромолекул полимера I в ТГФ может служить гауссов клубок, образованный цепями конечной длины, и найдено экспериментальное значение сегмента Куна, равное 30 А. При выборе модели для описания поведения макромолекул полимера II в ТГФ мы руководствовались теми же соображениями, что и в работе [7], поскольку так же, как и в работе [7]. параметр а в уравнении Марка — Куна — Хаувинка в 8-условпях не равнялся 0,5 (а=0,61) и заметно влияние растворителя па гидродинамические параметры (например, на [л]). Мы также воспользовались моделью клубка, образованного цепями конечной длины. Была построена зависимость М/[п] от √М и проведен расчет по уравнению [13]
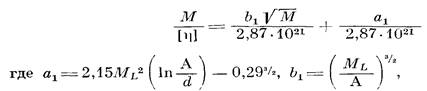
Так как для полиарилата III в Э-условиях значение а=0,5, в данном случае нами была использована модель гауссова непроницаемого клубка и значения А рассчитаны по уравнению [13]
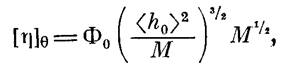
Результаты, приведенные в табл. 3, полностью подтвердили выводы, сделанные на основе данных машинного эксперимента. А именно введение в кислотную компоненту элементарного звена макромолекулы дополнительной фенильной группы привело к некоторому увеличению значения сегмента Куна с 30 А для полимера I до 40 А для полимера II, что свидетельствует о нарастании равновесной жесткости цепи. Введение дополнительной фталидной группировки (полимер III) понизило жесткость цепи и уменьшило значение сегмента Куна до 23 А. Кроме того, степени заторможенности о=УЛэД4т этих полимеров близки по значению и невелики, т. е. в растворе осуществляется большой набор возможных конформаций.
Таким образом, изменение строения кислотной компоненты элементарного звена в незначительной степени сказалось на равновесной жесткости макромолекулярной цепи; более заметно влияние этого фактора на термодинамический процесс растворения, и, следовательно, на структуру самого раствора.
ЛИТЕРАТУРА
1.Тагер А.А. // Высокомолек. соед. А. 1984. Т. 26. № 4. С. 659.
2.GoellК.В., BerryG.С. // J. PolymerSci. Phys. Ed. 1977. V. 15. № 3. P. 555.
3.Тагер А.А., Древалъ В.E.. Курбаналиев M., Луцкий М. С. Берковец Н. Б., Грановская И.М., Чарикова Т.А. // Высокомолек. соед. А. 1968. Т. 10. № 9. С. 2041.
4.Курбаналиев М., Тагер А.А., Древалъ В.Е. // Механика полимеров. 1968. № 2. С. 35.
5.Коршак В.В., Виноградова С.В., Салазкин С.Н. // Высокомолек. соед. 1962. Т. 4. № 3. С. 339.
6.Вайсбергер А., Проскауэр Э., Риддик Дж., Тупс Э. Органические растворители. М., 1958.
7.Коршак В.В., Павлова С.-С.А.. Дубровина Л.В., Кобак Н.Ю., Гладкова Е.А.Ц Высокомолек. соед. А. 1980. Т. 12. № 7. С. 1458.
8.Сердюк И. Н., Эскин В. Е. // Вестн. ЛГУ. 1970. Вып. 2. № 10. С. 57.
9.Flory P. Principles of Polymer Chemistry. N. Y., 1953. P. 532.
10.Готман В. Химия координационных соединений в неводных средах. М., 1971. С. 165.
11.Pavlova S.S. A., Timofeeva G.I., Pancratova L.//J. Polvmer Sci. PolymerPhys. Ed. 1980. V. 18. № 1. P. 1.
12.Флори П. Статистическая механика цепных молекул. М., 1971.
13.Рафиков С.Р., Будтов В.П., Монаков Ю.Б. Введение в физикохимию растворов полимеров. М., 1978.
|
