"Методы органического синтеза"
Введение
Органический синтез - раздел органической химии, в котором рассматриваются пути и методы искусственного создания органических соединений в лаборатории и промышленности. Широко применим в лабораторных условиях (главным образом для исследовательских целей) и в промышленности.
Успешное развитие органического синтеза началось после разработки теории химического строения и накопления сведений о химических свойствах органических соединений (2‑я пол. 19 в.). С этого времени органический синтез как основной источник новых органических соединений играет фундаментальную роль в становлении органической химии как науки и в ее дальнейшем развитии, обеспечивая постоянно расширяющийся круг изучаемых объектов. Развитие органического синтеза в 20 в., особенно в последние десятилетия, характеризуется все возрастающим вниманием к синтезу природных соединений и их аналогов, значительным укреплением методической базы (созданием надежных синтетических методов), началом создания самостоятельной теории органического синтеза. Осуществление синтеза сложнейших природных соединений (например хлорофилла, витамина В12
, биополимеров), создание материалов с необычными свойствами (например так называемые органические металлы
) показывает, что для современного органического синтеза практически не существует неразрешимых задач.
В реферате рассмотрены вопросы, касающиеся планирования органического синтеза, т.е. выбора оптимального пути получения соединения с заранее заданной структурой. Конкретные методы синтеза – образование новой связи С–С, введение функциональных групп и другое.
Обычно синтез целевого соединения осуществляют из относительно простых и доступных (т.е. выпускаемых промышленностью) исходных веществ. Как правило, при синтезе сложных веществ путь от исходных соединений к целевому разбивается на ряд этапов (стадий), на каждом из которых происходит образование одной – двух связей (фрагментов) будущей молекулы или подготовка к образованию таких связей.
Осуществление органического синтеза сопряжено с решением двух основных вопросов: 1) разработка общего плана синтеза, т.е. выбор оптимальных исходных соединений и последовательности стадий, ведущих кратчайшим путем к целевому продукту (стратегия синтеза); 2) выбор (или разработка новых) синтетических методов, обеспечивающих возможность построения необходимой связи в определенном месте собираемой молекулы (тактика синтеза).
Реклама

Основу тактики органического синтеза составляют различные синтетические методы, каждый из которых представляет собой стандартную совокупность одной или нескольких реакций и приемов выделения продуктов, которые обеспечивают возможность построения или разрыва определенного типа связи (или связей), необходимой для синтеза целевого соединения. Важные характеристики эффективного синтетического метода – общность (слабая зависимость результата от конкретных особенностей структуры исходных соединений), селективность (участие в основных реакциях метода лишь определенных функциональных групп) и высокие выходы продуктов. Типичным примером эффективного синтетического метода может служить синтез олефинов по Виттигу (реакции 1–3) из алкилгалогенидов и карбонильных соединений:

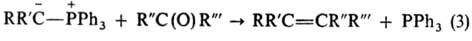
Основные методы органического синтеза можно разбить на три группы: 1) конструктивные, ведущие к образованию новых связей С–С, назначение которых – построение скелета будущей молекулы (например, реакция Гриньяра, реакция Фриделя-Крафтса, цикло – присоединение); 2) деструктивные, ведущие к разрыву определенных связей С–С с целью удаления той или иной группировки из молекулы после того, как ее роль в синтезе сыграна (например, декарбоксилирование, периодатное окисление диолов); 3) методы трансформации функциональных групп. Последнее важно для введения в молекулы исходных или промежуточных соединений функциональных групп и их защиты,требующихся для осуществления очередной конструктивной реакции, а на заключительных стадиях синтеза-для введения необходимых функциональных групп в целевое соединение.
Алкилирование и ацилирование
Реакция Шоттена-Баумана
, ацилирование спиртов или аминов хлорангидридами карбоновых к-т в присут. водного р-ра щелочи или соды (акцепторов образующегося НС1):
RCOC1 + HOR' + NaOH RCOOR' + NaCl + Н2
О RCOC1 + H2
NR' + Na2
CO3 RCOOR' + NaCl + Н2
О RCOC1 + H2
NR' + Na2
CO3 RCONHR' + NaCl + CO2
+ H2
O
RCONHR' + NaCl + CO2
+ H2
O
В качестве акцепторов НС1 применяют также NaHCO3
, CaO, MgO, CH3
COONa. Ацилирующими агентами обычно являются трудногидролизуемые хлорангидриды ароматических к-т (например, бензоилхлорид), а также хлорангидриды высших алифатических к-т (С10
-С18
). При ацилировании спиртов выход сложных эфиров повышается с понижением температуры реакции за счет снижения гидролиза хлорангидрида и отчасти эфира. Чтобы избежать местных перегревов, хлорангидрид прибавляют небольшими порциями к р-ру спирта в водной щелочи при эффективном перемешивании. Поскольку реакционная среда должна оставаться слабощелочной до конца р-ции, хлорангидрид и щелочь берут с 20–25%-ным избытком. Эти же правила применимы и для ацилирования аминов. Выходы 60–95%.
Ацилирование легкогидролизующимися хлорангидридами (СОС12
, AlkCOCl) проводят в инертных растворителях (диэтиловый эфир, хлороформ, бензол) в присутствии мелкоизмельченного порошка щелочи или соды.
Реклама

Аналогично спиртам в р-цию вступают тиолы:
RCOC1 + HSR' + NaOH RCOSR' + NaCl + Н2
О RCOSR' + NaCl + Н2
О
Ш.‑Б. р. используют для лабораторного и промышленного получения разложение сложных эфиров и амидов, например бензанилида C6
H5
NHCOC6
H5
. Реакцию применяют в аналитической практике для идентификации хлорангидридов в виде их анилидов и аминов в виде бензоильных производных.
Метод впервые применен К. Шоттеном в 1884 для ацилирования аминов и Э. Бауманом в 1886 для ацилирования спиртов.
Модификация Ш.‑Б. р. – метод Айнхорна, в к-ром вместо щелочи используют пиридин, служащий одновременно растворителем и акцептором НС1:
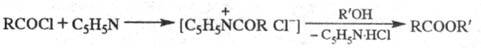
Метод находит широкое применение благодаря мягким условиям синтеза и высокой ацилирующей способности пиридиниевой соли. В ряде случаев вместо пиридина используют третичные амины, например (C2
H5
)3
N или (CH3
)2
NC6
H5
.
Алкилирование,
введение алкильной группы в молекулу органического соединения, а также получение алкильных производных химических элементов.
Наиб. часто в качестве алкилирующих агентов используют алкилгалогениды, алкены, эпоксисоединения, спирты, реже – альдегиды, кетоны, эфиры, сульфиды, диазоалканы.
Алкилирование изопарафиновых и ароматических углеводородов проводят: в жидкой фазе в инертном растворителе при температурах до 100 °С и давлении, необходимом для поддержания жидкофазного состояния; в паровой фазе с применением гетерогенных катализаторов при 200–350 °С и давлением 0,3–3,5 МПа. Например, алкилирование триметилметана бутеном осуществляют в жидкой фазе при 0–10 °С (кат. – Н25О4) или – 10 °С (HF), алкилирование бензола этиленом- в жидкой фазе при 90–100 °С (А1С13) или паровой фазе при 250 °С и давлением 3,5 МПа (BF3), алкилирование бензола пропиленом- в жидкой фазе при 50 °С и давлением 0,7 МПа (HF) или паровой фазе при 300 °С и давлением 0,3–1,0 МПа (H3P04/Si02).
О – алкилирование проводят при температурах не выше 100 С в воде или органических растворителях, например:
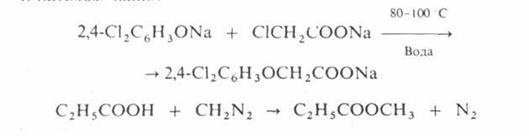
N – алкилирование аминов спиртами осуществляют в газовой фазе в присутствии кислотных катализаторов при 200–300 °С, напр.:
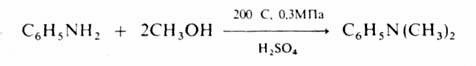
Получение алкильных производных металлов проводят в присутствии меди, например:

Алкилирование углеводородов сопровождается полиалкилированием, изомеризацией и полимеризацией. Так, при этилировании бензола по р-ции Фриделя – Крафтса, кроме этилбензола, образуются ди- и полиэтилбензолы. Полиалкилирование объясняется лучшей растворимостью в образующемся каталитическом комплексе алкилатов по сравнению с исходным в-вом. При использовании в качестве растворителя нитрометана идет преимущественное образование моноалкилпроизводных. Для увеличения выхода моноалкилпроизводных уменьшают мольное соотношение олефин: бензол, а также проводят рециркуляцию полиалкилпроизводных, в результате которой идет их деалкилирование.
Механизм алкилирования наиболее подробно изучен на примере С‑и О – алкилирования реакции с участием алкилгалогенидов, спиртов, сложных эфиров протекают следующим образом:
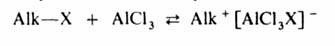 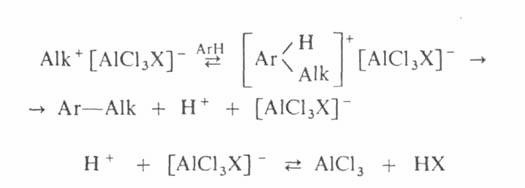
Поскольку третичные алкилгалогениды ионизируются легче всего, вторичные – труднее, а первичные практически не ионизируются, вероятность образования соответствующих карбкатионов уменьшается в том же ряду. Такое же влияние строения алкилирующего агента отмечено при О – алкилировании, которое в целом протекает как нуклеофильное замещение у насыщенного атома углерода:
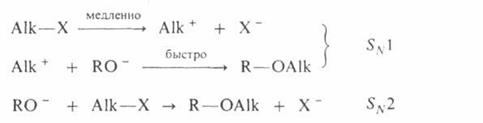
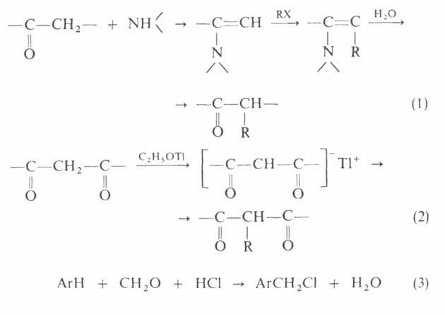
Помимо приведенных выше реакций, алкилирование применяют в лабораторной практике для получения алкильных производных карбонильных соединений, дикарбонильных соединения, при хлорметилировании ароматических углеводородов:
Алкилирование широко применяется в промышленности, в частности для получения алкилата, этилбензола, изопропилбензола, высших алкилбензолов.
Реакции конденсации
Исторически закрепившееся в органической химии название большой группы реакций различного характера. В более узком значении – внутри- и межмолекулярные процессы образования новой связи С–С в результате взаимодействия двух или более молекул органических соединений. Реакции конденсации можно разбить на след. группы: 1. Замещение атома или группы атомов с отщеплением простой неорганической или органической молекулы:

В качестве конденсирующих агентов используют в-ва, которые связывают отщепляющиеся соединения, образуют реакционноспособные промежуточные продукты или действуют как катализаторы. Реакции конденсации с отщеплением воды могут проходить по одной из след. схем:
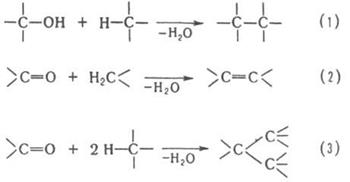
Схеме (1) соответствуют алкилирование ароматических и непредельных соединений спиртами, автоконденсация жирных спиртов, например:
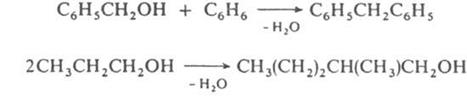
По схеме (2) протекают кротоновая конденсация и многочисленные родственные процессы, например Перкина реакция, Кнёвенагеля реакция и др.; по схеме (З) – многие синтезы ряда трифенилметана, например:
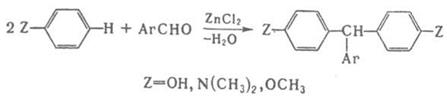
Отщепление воды катализируется обычно кислотами и основаниями, такими, как H2
SO4
, HCl, АlСl3
, ZnCl2
, NaOH, NaOR, NaNH2
, NaH, RNH2
. Некоторые реакции, сопровождающиеся образованием связи углерод–гетероатом или гетероатом–гетероатом, также относят к реакциям конденсации, например:
Под действием металлов реакции конденсации происходят с отщеплением атомов галогена от двух молекул орг. галогенида (Вюрца реакция, Ульмана реакция). Реакции конденсации с отщеплением водорода могут осуществляться пиролитически либо под действием окислителей, например:
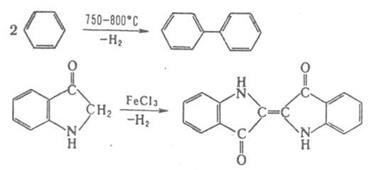
Ряд процессов реакции конденсации сопровождается отщеплением молекул орг. соединения, например спиртов. К этому типу принадлежат сложноэфирная конденсация, Клайзена конденсация, Дикмана реакция. Конденсирующие агенты – щелочные металлы, орг. и неорг. основания. Обычно к К. р. не относят этерификацию, переэтерификацию, алкилирование и ацилирование по гетероатомам, однако происходящие по этим схемам процессы образования полимеров называют поликонденсацией. 2. Присоединение молекулы органического соединения по кратной связи другой молекулы:
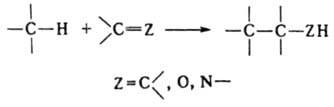
Сюда относят, например, многочисленные случаи альдольной конденсации, зачастую представляющей собой предварительную стадию кротоновой конденсации, Михаэля реакцию, бензоиновую и ацилоиновую конденсации, диеновый синтез, а также реакции гидро- и карбометаллирования олефинов и ацетиленов.
Диазотирование
Способ получения ароматических диазосоединений, заключающийся обычно в действии NaNO2
на первичные ароматические амины в присутствии минеральной к-ты НХ:
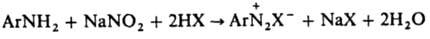
Диазотирование проводят в воде, концентрированных кислотах, реже – в неводных средах. Поскольку реакция экзотермична, а диазосоединения при нагревании легко разлагаются, реакционная смесь обычно охлаждают, поддерживая температуру в интервале 0–10 °С. При недостатке кислоты могут образовываться диазоамино- и аминоазосоединения. Производные о-аминонафтолов при диазотировании окисляются; для предотвращения этого в реакционную смесь добавляют соли Сu или Zn. Механизм диазотирования включает нитрозирование свободного амина с последующим отщеплением Н2
О от катиона N‑нитрозаммония (I) или ОН –
от N‑нитрозамина (II):
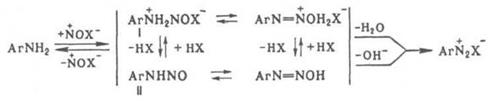
Нитрозирующий агент NOX образуется по р-ции: NO2
-
+ + 2Н+
+ Х-
D NOX + Н2
O, где X = ОН, ОС(О) CН3
, OSO3
H, NO2
, Hal и др. (X расположены в порядке возрастания активности NOX). Наиболее активный агент – свободный нитрозоний – катион NO+
; он образуется только в концентрированной серной или хлорной к-те. Если NOX образуется быстрее, чем катион N – нитрозаммония, скорость диазотирования зависит от концентрации амина. Чем ниже кислотность среды, тем выше концентрация NO2
-
и ОН-
, а следовательно, и концентрация малоактивных частиц N2
O3
и HNO2
, в результате чего скорость диазотирования должна снижаться. Однако одновременно увеличивается концентрация свободного амина, что приводит к увеличению скорости диазотирования. С увеличением кислотности среды, как правило, увеличивается концентрация наиболее активных NOX, однако уменьшается концентрация свободного амина, что приводит к снижению скорости диазотирования. Поэтому в слабокислой среде диазотируют более основные амины, в сильнокислой – менее основные, в концентрированной H2
SO4
с помощью нитрозилсерной к-ты – амины крайне низкой основности (например, полинитроанилины). Чтобы увеличить скорость последней р-ции, среду разбавляют ледяной СН3
СООН, сдвигая равновесие в сторону образования свободного амина. При диазотировании обычно к р-ру или мелкодисперсной суспензии соли амина в к-те прибавляют NaNO2
, взятый с небольшим избытком. При использовании плохо растворимых аминосульфокислот к слабощелочному р-ру амина, содержащему NaNO2
, прибавляют соляную к-ту. Для выделения галогенидов диазония процесс ведут в абсолютном спирте или ледяной СН3
СООН, используя http://www.xumuk.ru/encyklopedia/909.html водородные соли амина и в качестве диазотирующего агента – алкилнитриты. Для контроля р-ции в промышленности используют анализаторы с электро – химической индикацией избыточной HNO2
. Анализатор связан с автоматическим дозиметром, регулирующим прибавление к-ты, NaNO2
и амина таким образом, чтобы не возникал избыток нитрозирующего агента. диазотирования – первая стадия синтеза азокрасителей, а также р-ций Зандмейера, Гомберга, Шимана, Гаттермана, Несмеянова, Барта, Меервейна. Диазотирование открыто П. Гриссом в 1858.
Нитрование
Введение нитрогруппы – NO2
в молекулы органических соединений. Может проходить по электрофильному, нуклеофильному и радикальному механизмам; активные частицы в этих реакциях – соответственно катион нитрония NO2
, нитрит-ион NO2
и радикал NO2
. Нитрование может осуществляться по атомам С, N, О замещением атома водорода (прямое нитрование) или других функциональных групп (заместительное нитрование) либо в результате присоединения группы NO2
по кратной связи.
Электрофильное нитрование.
Среди электрофильных нитрующих агентов доминирующее положение занимает HNO3
. Безводная и конц. HNO3
способны к самопротонированию: 2HNO3
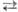 [Н2
NО3
]+
+ NO3
- [Н2
NО3
]+
+ NO3
-
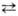 NО2
+
+ NO-
3
+ H2
O. Присутствие воды снижает концентрацию NO+
2
и в 93 – 95%-ной HNO3
спектрофотометрически он уже не обнаруживается. Для увеличения нитрующей активности HNO3
используют ее смеси с H2
SO4
или олеумом, к-рые генерируют NO2
, связывая воду: NО2
+
+ NO-
3
+ H2
O. Присутствие воды снижает концентрацию NO+
2
и в 93 – 95%-ной HNO3
спектрофотометрически он уже не обнаруживается. Для увеличения нитрующей активности HNO3
используют ее смеси с H2
SO4
или олеумом, к-рые генерируют NO2
, связывая воду:
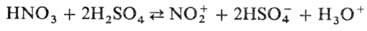
В безводной H2
SO4
при содержании HNO3
меньше 10% равновесие полностью сдвинуто вправо. Применяют также комбинации HNO3
, разложение оксидов азота и органических нитратов с кислотами Льюиса (АlСl3
, ZnCl2
, BF3
и др.); сильным нитрующим действием обладает смесь HNO3
с (СН3
СО)2
О благодаря образованию ацетилнитрата и N2
O5
(последний при содержании в смеси более 90% HNO3
полностью диссоциирует на NO+
2
и NO-
3
); перспективны также смеси HNO3
с безводным SO3
или N2
O5
. Вместо HNO3
можно применять ее соли, однако в промышленности такой метод не используют из-за осложнения процесса регенерации отработанных к-т. В случае слабой взаимной р-римости нитрующего агента и субстрата, а также для уменьшения побочных процессов нитрование проводят в органических р-рителях, например нитрометане, сульфолане, уксусной к-те; полярные р-рители способствуют диссоциации [H2
NO3
]+
и тем самым увеличивают концентрацию NO2
.
В лабораторной практике широко используют апротонные нитрующие агенты (нитраты, соли нитрония, полинитросоед. и др.), активность которых в реакциях электрофильного нитрования увеличивается в ряду: AlkONO2
< (CH3
)2
C(CN) ONO2
< < RC(N02
)3
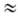 RN(N02
)2
< NO2
F < CH3
COONO2
< < N2
O5
< NO2
+
X-
. RN(N02
)2
< NO2
F < CH3
COONO2
< < N2
O5
< NO2
+
X-
.
Субстратами для электрофильного нитрования служат ароматические и гетероциклические соединения, олефины, относительно сильные СН – кислоты, амины, спирты.
Нитрование ароматического соединения протекает по схеме:
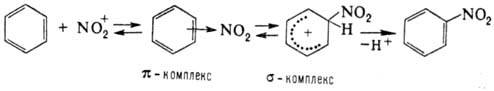
Возможно также образование s‑комплекса, в котором группа NO2
связана с атомом углерода кольца, несущим заместитель. Соединения с электронодопорными заместителями более реакционноспособны и нитруются в орто- и пара-положения, а с электроноакцепторными – в мета-положение. В промышленности для нитрования ароматических соединений применяют в основном смесь HNO3
и H2
SO4
(выход нитропродуктов ~ 90–95%). Основная побочная р-ция – окисление, приводящее, как правило, к деструкции ароматического кольца. В зависимости от реакционной способности субстрата условия нитрования варьируют в широких пределах – от водной HNO3
при 0 °С (обязательно присутствие оксидов азота) до дымящей HNO3
в олеуме при повышенных температурах. При низких температурах с высокой скоростью протекает нитрование ароматических соединений солями нитрония; при этом часто лимитирующая стадия-скорость растворения соли нитрония. Используют также заместительное нитрование – замещение сульфо-, диазо- и др. функциональных групп. Этим приемом пользуются, в частности, в случаях, когда невозможно прямое нитрование. Нитрование олефинов апротонными нитрующими агентами в зависимости от условий и строения реагентов может идти по разным направлениям, включая отщепление Н+
, присоединение элементов р-рителя и противоиона, полимеризацию и др., например:
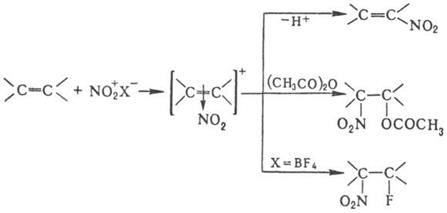
При нитровании олефинов тетранитрометаном в зависимости от строения олефина образуются либо алифатическое полинитро – соединение, либо производные изоксазолидина, например:

Некоторые СН – кислоты при нитровании образуют анионы соответствующих нитросоединений; например, при действии на флуорен этил – нитрата в присутствии С, Н5
ОК образуется К – соль 9‑нитро-флуорена, примером нитрования карбанионов может служить также превращение солей моно- и динитросоединений соответственно в геминальные ди- и тринитропроизводные при действии FNO2
. Соединения с активированной метиленовой группой можно нитровать и в кислых условиях; например, при обработке диэтилмалоната HNO3
образуется нитродиэтилмалонат, нитрование в аналогичных условиях 1,3 – индандиона с последующим щелочным гидролизом образующегося a – нитрокетона – удобный метод синтеза первичных нитроалканов:
Электрофильное нитрование аминов в отличие от нитрования по атому С – обратимый процесс и протекает по схеме:
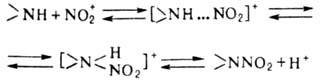
В промышленности нитрование аминов проводят кислыми нитрующими агентами (конц. HNO3
или ее смесями с H2
SO4
, уксусной к-той или ангидридом). Слабоосновные амины и амиды нитруются с высокими выходами. Высокоосновные амины (первичные и вторичные), протонирующая форма которых не реагирует с NO2
+
, превращают либо в амиды, которые нитруют и затем снимают защитную ацильную группу щелочным гидролизом, либо в N‑хлорамины; в последнем случае нитрование проводят в присутствии катализаторов (НСl, ZnCl2
).
Нитрование третичных аминов конц. HNO3
или ее смесью с уксусным ангидридом сопровождается разрывом связи С– N (такой тип нитрирования называется нитролизом). Эту реакцию широко используют в промышленности, например для получения гексагена и октогена из уротропина. Жирно – ароматические амины типа ArNHR часто нитруются в ядро, что происходит в результате непосредственного нитрования по атому С или перегруппировки N‑нитропроизводного; при этом группа NO2
вступает в ортo – положение к аминной функции. В ряде случаев нитрования по атому N проводят через стадию образования соли. Для этого амин обрабатывают разб. HNO3
и на образовавшийся нитрат действуют конц. HNO3
или уксусным ангидридом:
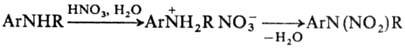
В лабораторных условиях заместительное нитрование ацетамидов, сульфамидов, уретанов, имидов или их солей проводят в апротонной среде апротонными нитрующими агентами, например солями нитрония:
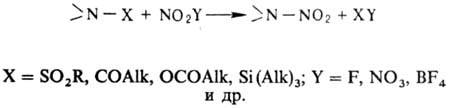
Из первичных аминов можно синтезировать N, N‑дини-троамины, которые, в свою очередь, являются нитрующими агентами.
Спирты нитруют любыми нитрующими агентами, содержащими NO+
2
(в кислых средах р-ция обратима), например: RCH2
OH + NO2
+
X-
 RCH2
ONO2
+ НХ. RCH2
ONO2
+ НХ.
Нуклеофильное нитрование
осуществляют солями HNO2
:
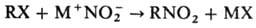
В реакцию вступают алкилгалогениды, в основном бромиды и иодиды, a‑галогенкарбоновые к-ты и их эфиры, алкилсульфаты. В качестве нитрующих агентов используют нитриты щелочных металлов в апротонных диполярных р-рителях или проводят нитрование в присутствии краун – эфиров. Побочные продукты реакции – органические нитриты, что связано с двойственной реакционной способностью NO-
2
. Реакцию используют для получения алифатических нитросоединений.
Радикальное нитрование.
Характерно в основном для парафинов и олефинов. Источником NO.
2
служат HNO.
3
и оксиды азота. Нитрование парафинов проводят разб. HNO3
под давлением при повышенной температуре (Коновалова реакция). Р-ция нитрования протекает по схеме:
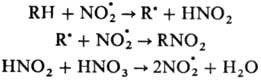
Наряду с нитрованием идет также процесс окисления, связанный с взаимодействием NO.
2
с орг. радикалом по атому кислорода. Наиболее легко протекает нитрование по третичным атомам углерода, трудно – по первичным. В промышленности нитропарафины получают жидкофазным и парофазным нитрованием смеси парафинов. Жидко-фазное нитрование проводят HNO3
при нормальном или повышенном давлении и температуре выше 180 °С, или оксидами азота при давлении 2–4,5 МПа, 150–220 °С, время контакта ~15 с. В этих условиях линейные углеводороды нитруются быстрее, чем их разветвленные изомеры. Парофазное нитрование (метод Хэсса) осуществляют HNO3
при давлении 0,7–1,0 МПа, 400–500 °С, время контакта ~ 1 с. Побочные процессы – деструкция углеводородной цепи и окисление. Эти методы используют также для нитрования алифатических боковых цепей жирно – ароматических соединений (р-цию проводят в присутствии катализаторов – О2
, О3
, галогенов и др.),
Нитрование непредельных соединений HNO3
приводит к формальному замещению атома водорода у sp2
-гибридизованного атома углерода на группу NO2
. Условия нитрования зависят от строения непредельных соединений. Обычно применяют 70–80%-ную HNO3
или разб. HNO3
в присутствии оксидов азота.
Галогенирование (галоидирование)
Введение галогена в молекулу орг. соединения. Осуществляют путем р-ций замещения (заместительное галогенирования) или присоединения (присоединительное галогенирование).
Заместительное галогенирование. При действии галогенов на насыщ. углеводороды (металепсия) процесс протекает при инициировании светом по свободнорадикальному цепному механизму, например:
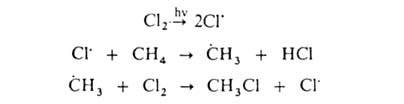
По свободнорадикальному механизму идет также галогенирование углеводородных цепей жирноароматических соединений. В присутствии к-т Льюиса р-ция протекает по электрофильному механизму, напр.:

Галогенирование алифатических карбоновых к-т в -положение проводят с помощью С12
или Вг2
в присутствии красного Р (Гелля-Фолъгарда-Зелинского реакция). Замещение -положение проводят с помощью С12
или Вг2
в присутствии красного Р (Гелля-Фолъгарда-Зелинского реакция). Замещение водородных атомов в алифатических и жирноароматических карбонильных соединений идет через присоединение галогена к фенольной форме, например: водородных атомов в алифатических и жирноароматических карбонильных соединений идет через присоединение галогена к фенольной форме, например:
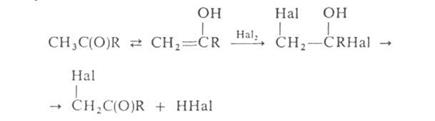
Действием N – галогенамидов, главным образом N – бромсукцинимида, в присутствии пероксидов осуществляют свободнорадикальное галогенирование олефинов, жирноароматических и гетероароматических соединений по метальной или метиленовой группе, соседней с двойной связью или циклом (Воля – Циглера реакция).
Замещение атомов Н на F с образованием полифторзамешенных соединений проводят путем электрохимеского фторирования в безводном HF (р-ция Саймонса), действием CoF3
и др.
Заместительное галогенирование в ядро ароматических и гетероароматических соединений протекает по механизму электрофильного замещения; обычно его осуществляют с использованием катализаторов (гл. обр. апротонных или протонных к-т), например:
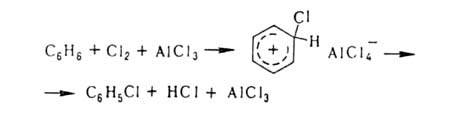
Если в ядре этих соединений присутствуют пассивирующие заместители, процесс можно проводить действием катиона галогена, образующегося из молекулярного галогена и соли Ag в среде сильной протонной к-ты (р-ция Биркенбаха-Губо-Уотерса), например:
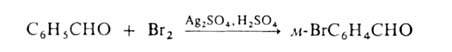
Замещение на галоген атомов, отличных от водорода, или группы атомов осуществляется чаще всего по нуклеофильному механизму. В алифатических соединениях для замены атомов галогенов на иод используют Nal (Финкелъштапна реакция), а на фтор-SbF3
(р-ция Свартса). Группы ОН замещают на хлор или бром действием соответствующих галогеноводородов, тригалогенидов или оксигалогенидов фосфора, а также тионилгалогенидов, а на фтор – действием диэтил – 1,1,2 – трифтор‑2‑хлорэтиламина или SF4
. Карбоксильную группу замещают на С1, Вг или I действием на серебряные соли карбоновых к-т соответствующего галогена (Бородина – Хунсдиккера реакция). Заменой карбонильного кислорода в альдегидах или кетонах на галоген (например, с помощью РС15
, PBr5
, SF4
, MoF6
) получают геминальные галогензамещенные.
В ароматическом ряду для получения галогензамсщеиных используют замену групп NH2
на С1, Вr или I каталитическим разложением соответствующих солей диазония в присутствии порошка Сu (Гаттермана – Коха реакция) или действием солей Сu (Зандмепера реакция), а на F‑разложением гидрофторидов диазония (Шимана реакция). Для галогенирования ароматические и гетероароматические соединения используют также р-цию замещения (в т.ч. обмен галогенов), протекающую по механизму присоединения-отщепления с промежуточным образованием анионных комплексов, напр.: комплексов, напр.:
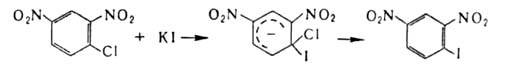
Присоединительное галогенирование.
К ароматическим и гетероароматическим соединениям галоген присоединяется, как правило, по радикальному механизму под действием света или при нагревании, например:
Если цикл активирован, р-ция может протекать по ионному механизму, к-рый включает стадию присоединения аниона галогена к промежуточно образующемуся в процессе электрофильного замещения комплексу, например: комплексу, например:
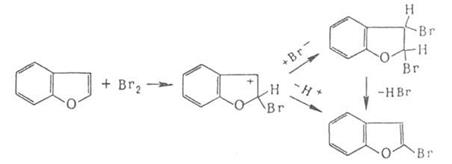
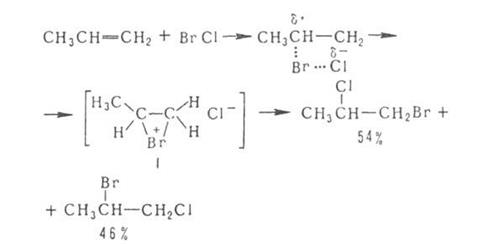
Присоединение галогенов по кратной связи происходит по электрофильному или радикальному механизму. Его можно осуществлять действием галогсноводородов, межгалогенных соединений или гипогалогенитов. В случае электрофильного присоединения может нарушаться правило Марковникова, что обусловлено образованием промежуточного мостикового катиона, например:
Сульфирование (сульфонирование).
Введение сульфо – группы SO2
OH в молекулу орг. соединения; в широком смысле сульфирование – введение группы SO2
X (X = ОН, ONa, OAlk, OAr, Hal, NAlk2
и т.п.). О введении группы SO3
H с образованием связей О–S (О – сульфирование, сульфатирование, сульфоэтерификация).
Процесс, обратный сульфированию (удаление группы SO2
X из молекулы орг. соединения), называется десульфированием (десульфонированием). Сульфирование осуществляют прямым путем с использованием сульфирующих агентов либо косвенным путем, например введением сульфогруппы в составе сульфоалкильных фрагментов (СН2
)n
SО2
Х. Сульфирующие агенты: H2
SO4
, SO3
и его комплексы с орг. соединениями (эфирами, третичными аминами и фосфинами, амидами карбоновых кислот, триалкилфосфатами и др.), олеум, SOCl2
, галогенсульфонрвые и сульфаминовые к-ты, диалкилсульфаты, ацилсульфаты.
Сульфирование ароматических углеводородов протекает по механизму электрофильного замещения:
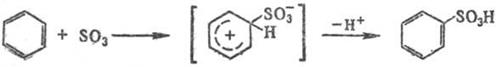
Реакцию осуществляют как в паровой, так и в жидкой фазе (р-рители: SO2
, СС14
, хладоны и т.п.). При сульфировании серной к-той для смещения равновесия вправо применяют избыток к-ты или связывают воду добавлением олеума, азеотропной отгонкой и т.п.
Соединение с электронодонорными заместителями более реакционноспособны и сульфируются преимущественно в орто- и пара-положения; соединения с электроноакцепторными заместителями- в мета-положение. В большинстве случаев при сульфировании замещенных бензолов образуются смеси изомеров, соотношение к-рых зависит от природы заместителя, сульфирующего реагента и условий р-ций (концентрации реагентов, т-ры, р-рителя, наличия катализаторов и т.д.). Путем подбора оптимальных условий возможно селективное сульфирование. Так, сульфирование толуола серной к-той при 20 °С приводит к равным кол-вам о- и n‑толуолсульфокислот, а при, использовании SO3
в тех же условиях – исключительно к n‑изомеру; при сульфировании фенола на холоду преимущественно образуется о-фгнолсульфокислота, тогда как при 100 °С-n‑фенолсульфокислота. Как правило, подобные различия обусловлены превращением одних изомеров в другие, термодинамически более стабильные, благодаря изомеризации или обратимости сульфирования. Например, нафталин при температурах ниже 100 °С первоначально образует a‑наф-талинсульфокислоту, к-рая во времени превращается в b‑изомер в результате последовательного десульфирования – ресульфирования. Сульфирование при 160 °С приводит исключительно к b‑нафталинсульфокислоте.
Для сульфирования гетероциклических соединений (фуран, пиррол, тиофен, индол и др.) используют комплексы SO3
с диоксаном или пиридином. Эти же реагенты применяют для сульфирования алифатических соединений, содержащих сильные электроноакцепторные группы; при этом образуются, как правило, a‑сульфопроизводные:
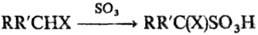
X = СНО, COR:, COOH, CN, NO2
, SO3
H идр.
Повышение СН-кислотности алифатических соединений способствует тому, что последующее сульфирование протекает более однозначно, чем моносульфирование. Например, ацетальдегид и уксусная к-та с высоким выходом образуют соответствующие да- и трисульфосоединения:
СН3
СНО + 2SO3
·Диоксан: (HSO3
)2
CHCHO
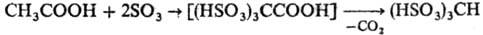
Легко реагируют с SO3
и его комплексами алкены, которые образуют в зависимости от структуры и условий b- или d‑сультоны (см. Сулътоны) либо a, b- или b, g‑ненасыщ. сульфокислоты, а также b‑гидроксисульфокис-лоты, напр.:
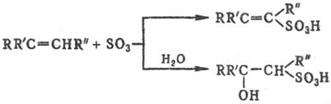
Прямое С. алканов протекает с трудом и сопровождается, как правило, окислением. Подобные р-ции редко используют для препаративных целей, но находят им практич. применение для С. полимеров, напр. полиэтилена, с целью их модификации. Значительно легче сульфируются углеводороды при совместном действии SO2
и О2
(сульфо-окисление), а также SO2
и Сl2
(сульфохлорирование). Обе р-ции имеют радикальный характер и инициируются пе-роксидами, УФ или g‑облучением:
Эти процессы имеют важное значение в пром. произ-ве ПАВ.
Для С. орг. соединений широко используют H2
SO3
и ее производные. Гидросульфиты присоединяются к альдегидам и кетонам (р-ция 1), а также в условиях радикального инициирования к алкенам и алкинам (2,3); взаимод. с трифенил- и три (n‑толил) карбинолами (4); реагируют с оксиранами, тииранами с раскрытием кольца (5):
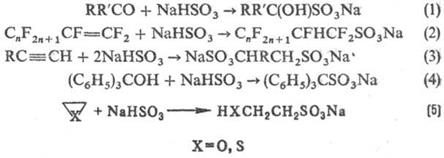
Диазоалканы сульфируют SO2
в присутствии воды, спиртов, тиолов и аминов (6), последоват. действие SO2
и галогена на реактивы Гриньяра приводит к сульфонил-галогенидам (7):
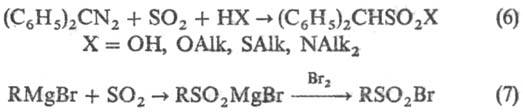
К косвенным методам С. относят сульфометилирование, сульфоэтилирование и т.д., напр.:
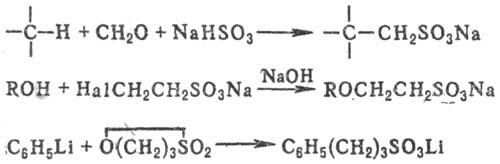
Амины. N‑ОКСИДЫ (N‑окиси аминов).
Производные третичных аминов и гетероароматич. соединений, содержащие семиполярную связь –О-
Большинство хорошо раств. в воде, ограниченно-в неполярных орг. р-рителях. Образуют с к-тами устойчивые кристаллич. соли –О-
Большинство хорошо раств. в воде, ограниченно-в неполярных орг. р-рителях. Образуют с к-тами устойчивые кристаллич. соли
R3 OHX-
Расположение четырех заместителей у атома азота тетраэдрическое.
OHX-
Расположение четырех заместителей у атома азота тетраэдрическое.
Свойства n‑оксидов аминов
| Показатель Т. пл., °С |
(СН3
Ь -O-
212,0 -O-
212,0 |
С6
Н5
(СН3
)2 -O
-O |
С5
Н5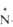 -O-
65,6
-O-
65,6 |
| Т. кип., °С |
– |
154,0 |
100* |
 *10-30
, Кл-м *10-30
, Кл-м |
16,7 |
16,15 |
14,11 |
| рКа
|
4,65 |
4,21 |
1,90 |
| Потенциал полуволны восстановления, В |
-0,456 |
-0,705 |
– 1,278 |
v( ->O-
), см-1 ->O-
), см-1
|
930–970 |
– |
1230–1320 |
А.о.-более слабые основания, чем исходные амины. Их основные св-ва определяются отрицат. зарядом на кислороде, по к-рому происходит как присоединение протона, так и алкилирование. При действии арилгалогенидов образуются соли тетразамещенного гидроксиаммония, разлагающиеся при обработке щелочью:
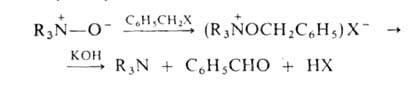
При ацилировании А.о. происходят след. превращения:
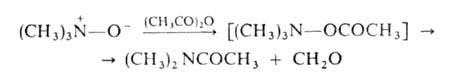
При нагр. алифатич. А. о. образуют замещенные гидроксиламины (перегруппировка Майзенхаймера) или олефины (перегруппировка Коупа), напр.:
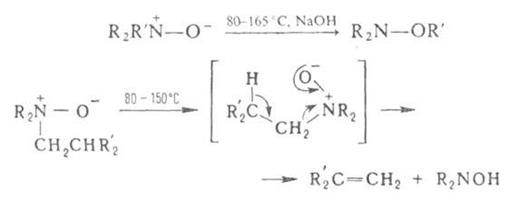
где R – Alk, Ar; R' = СН2
С6
Н5
, СН(С6
Н5
)2
, СН2
СН=СН2
.
А. о. восстанавливаются до аминов гидрированием на Ni или Pd, а также действием производных трехвалентного фосфора, напр. (С6
Н5
bР- В электронной системе цикла гетероциклич. А. о. группа электронной системе цикла гетероциклич. А. о. группа –О –
может играть роль как донора, так и акцептора электронов. Поэтому А. о. такого типа вступают в р-ции электроф. и нуклеоф. замещения легче, чем соответствующие амины. Ацилирование гетероциклич. А. о. происходит след. образом: –О –
может играть роль как донора, так и акцептора электронов. Поэтому А. о. такого типа вступают в р-ции электроф. и нуклеоф. замещения легче, чем соответствующие амины. Ацилирование гетероциклич. А. о. происходит след. образом:
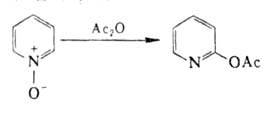
N‑Оксиды пиридина и его гомологов, производных хинолина нитруются до 4‑нитропроизводных. При р-ции с РОС13
, РС15
, SOC12
образуются 1- и 4‑хлорпроизводные, с ангидридами и галогенангидридами к-т‑2‑ацилоксипроизводные с металлоорг. соед. – 2‑алкилпроизводные, с анионом CN-
в присут. хлористого бензоила‑4‑цианпиридин.
Общий метод получения А.о.-окисление третичных аминов действием Н2
О2
в нейтральной (алифатич. амины) или кислой (ароматич. амины) средах, реже – озоном или надкислотами (азотистые гетероциклы). Применяется также исчерпывающее алкилирование гидроксиламина и его производных, циклизация нитро- и нитрозосоединений. Методы анализа А. о. основаны на восстановлении группы – О –
(потенциометрия). – О –
(потенциометрия).
Алифатич. А.о.-ПАВ в космети-ке и парфюмерии, коагулирующие и желатинизирующие агенты, ингибиторы полимеризации, р-рители целлюлозы. Нек-рые А. о. обладают противомикробной и противогрибковой активностью.
|
