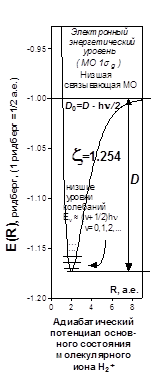.
Молекулярные интегралы и формула энергетические уровни:
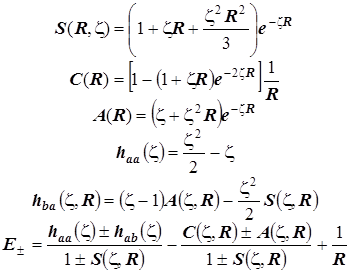
Эти формулы удобны для графического исследования уровней МО с помощью компьютера.
Наконец, для проверки физической корректности расчётов и положенных в их основу схем проанализируем предельные значения интегралов и уровней энергии МО:
Пределы интегралов (Квази-ион He+) Пределы электронной энергии
Можно видеть, что с физической точки зрения расчёт совершенно верно предсказывает пределы изменения электронной энергии системы в электростатическом поле ядер в гипотетическом процессе их сближения от бесконечного удаления до гипотетического слияния. Так подтверждается корректность теории, и это особенно важно, поскольку при её построении было использовано значительное количество непростых приближений.
В простейшей модели без оптимизации базисной АО получаем :
Показатель экспоненты в АО фиксирован и равен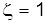
Все выводимые ниже выражения легко получаются из более общих выражений при
Интегралы существенно упрощаются и получаются следующие выражения:
1) Уровень исходной базисной АО 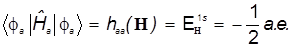
2) Интеграл перекрывания:
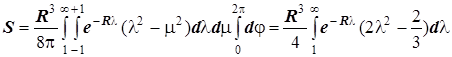 . .
Интегрируя по частям, получаем
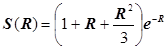 , ,
3) Кулоновский интеграл:
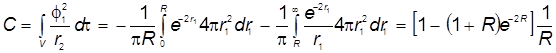 , ,
4) Резонансный интеграл:
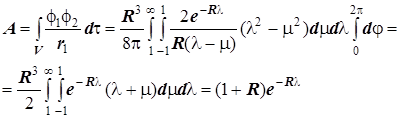
Отсюда получаются энергетические уровни МО в виде:
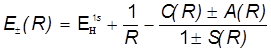 . .
Этот простой подход был исторически первым на пути построения квантово-механической теории валентности. Несмотря на свою ограниченность, он позволил на качественном уровне понять и происхождение электронного облака в межъядерной области, и природу устойчивости простейшей молекулярной системы. В количественном отношении этот примитивный подход очень слаб, и, вроде бы, не идёт ни в какое в сравнение с уточнёнными расчётами. НО...!!!
Самый трудный шаг на неизведанном и полном неясностей пути создания ранее не существовавшей теории всегда первый. Автор этого учебного текста наблюдал триумфальное развитие квантовой химии с середины 60-х годов по настоящее время (январь 1999 года) и застал переход от её исходного состояния к уже современному этапу и видел ещё слегка недоверчивое, изумлённое отношение химиков-синтетиков - людей, вообще-то весьма прагматично и дерзко мыслящих о веществе, к необычному ещё в то время варианту теории валентности, которая властно и как бы играючи вытесняла вариант привычной с 19 века качественной теории Бутлерова, оперирующей валентными штрихами с её причудливым нагромождением дополнительных конструкций. Оказалось, что не только качественно, но и количественно можно легко и точно объяснять и предсказывать спектрально наблюдаемые свойства молекул. Автор со студенческих лет хорошо помнит многочисленные дискуссии о сравнительных достоинствах и недостатках методов МО ЛКАО и ВС ЛКАО. Где-то сейчас метод ВС...?!!
Реклама

Бесспорным фаворитом теории валентности стал метод МО ЛКАО, идеально приспособленный к алгоритмам современной вычислительной математики и компьютерной техники.
Сейчас уже совершенно ясно, что теория ЛКАО МО была настоящей идейной революцией. В её основу положено одноэлектронное приближение. Молекулярный ион водорода был первой и простейшей системой, на примере которой было понято и теоретически изучено физическое происхождение феномена валентности.
Необходимые молекулярные интегралы принимают вид
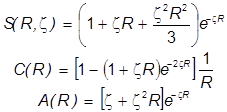 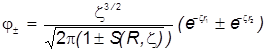 . .
Выражая локальные переменные (r
1
,
r
2
) через единые декартовы координаты , запишем выражение МО в виде:
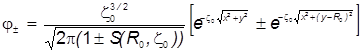 . .
Оптимизированные параметры 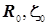 отвечают абсолютному минимуму целевой функции - полной энергии связывающей МО, определяемой в зависимости от двух переменных: межъядерного расстояния и эффективного заряда ядра - показателя экспоненты в формуле базисной АО. Энергетические уровни передаются формулой, на первый взгляд того же вида, что и в расчётах с одним варьируемым параметромR
: отвечают абсолютному минимуму целевой функции - полной энергии связывающей МО, определяемой в зависимости от двух переменных: межъядерного расстояния и эффективного заряда ядра - показателя экспоненты в формуле базисной АО. Энергетические уровни передаются формулой, на первый взгляд того же вида, что и в расчётах с одним варьируемым параметромR
:
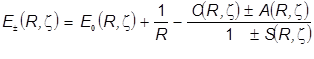 . .
Однако весьма существенное качественное отличие этой формулы состоит в том, что расчёт с двумя варьируемыми параметрами R
, z состоит в том, что 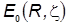 в общем случае является довольно сложной функцией обеих переменных, и лишь его предел переходит в величину E1
s
(H): в общем случае является довольно сложной функцией обеих переменных, и лишь его предел переходит в величину E1
s
(H):
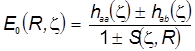 , ,

Оптимизация энергетического уровня за счёт дополнительного варьирования показателя экспоненты приводит к намного лучшему согласию с экспериментом.
В первых оригинальных работах в 20-х годах эти три характеристики основного состояния были рассчитаны с применением точной аналитической расчётной процедуры (Хиллераас). В настоящее время мощность вычислительной техники такова, что вся эта вычислительная процедура легко моделируется численными методами и реально доступный объём вычислений таков, что без особых проблем достигается любое графическое сопровождение расчётов, и те же самые результаты легко получаются в рамках прямого числового расчёта функции . На её пространственном графике, называемом потенциальной поверхностью, наблюдается искомый абсолютный минимум. Его координаты следующие: . На её пространственном графике, называемом потенциальной поверхностью, наблюдается искомый абсолютный минимум. Его координаты следующие:
Реклама

|
|
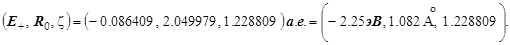
| Варьирование эффективных зарядов в базисных атомных орбиталях оказалось очень эффективным и гибким способом резкого улучшения расчёта молекулярного строения. Поэтому оно заняло очень важное место среди специфических приёмов современной квантовой химии. Есть и другие дополнительные способы улучшения базисных АО, но всё это означает лишь то, что в расчётах молекул атомные орбитали являются не более, чем удачными математическими базисными единицами разложений сложных функций МО в ряды вида ЛКАО. Рассматривать же АО в молекуле в самостоятельной физической роли нецелесообразно, хотя такой соблазн велик, и среди химиков ещё лет 20 назад был очень распространён.
|
|
График функции
представляет собой поверхность. Рассматривая переход системы в минимум энергии вдоль одного лишь межъядерного расстояния, не следует забывать о сопутствующем изменении и второй переменной - показателя экспоненты базисной АО. Мысленное сближение частиц протекает в условном энергетическом минимуме адиабатического потенциала и завершается достижением точки абсолютного минимума. Условный минимум на поверхности энергии представляет собою пространственную кривую, а его проекция на координатной плоскости это плоская кривая, которую называют координатой реакции исследуемого процесса. В этом процессе образование молекулярной системы формально является лишь промежуточной стадией.
Применяя графические процессоры для современных персональных компьютеров (MATHCAD
PLUS/PENTIUM 2,3,4), можно проиллюстрировать все вычисления. Наглядные пространственные графики на рис. изображают адиабатические потенциалы основного и первого разрыхляющего одноэлектронных уровней E
±
(z,R
).

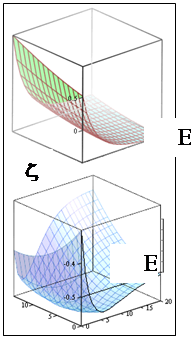
| Рис. 1. Зависимости от межъядерного расстояния одноэлектронных молекулярных интегралов: перекрывания
S
, кулоновского
C
, обменного
A
.
|
|
|
|
|
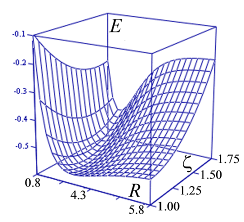
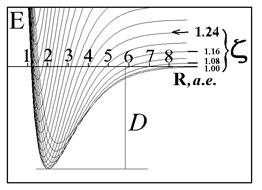
| Рис.4а. Фрагмент адиабатического потенциала
E
(
R
,
z
) молекулярного иона
H
2
+
в области минимума.
|
|
Рис.3. Оптимизиро-ванная энергетичес-кая кривая низшего связывающего уров-ня МО и низшие уровни колебаний
у молекулярного
иона водорода
H
2
+
|
|
| Рис.2. Слагаемые и ре-зультирующая кривая энергии основного элект-ронного состояния моле-кулярного иона водорода
H
2
+
|
|
| Рис.4. Варьирование экспоненты базисной АО: сечения адиабати-ческого потенциала для разных
z
и определение минимума энергии в основном состоянии иона
H
2
+
.
|
|
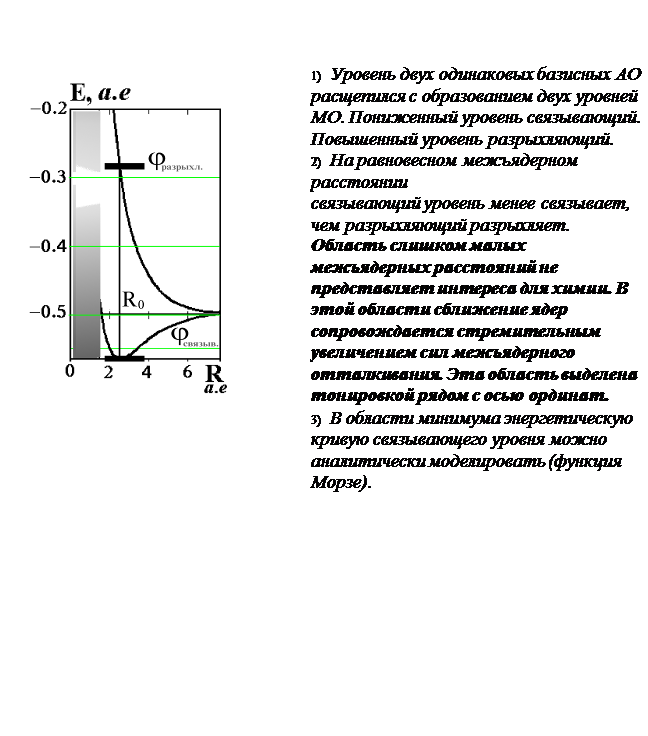
Химическая связь есть результат баланса электростатических сил . Энергия связи представляет собой малую разность больших величин.
|
|
Для анализа свойств
двухцентровой химической связи удобно выделить результирующую энергетическую кривую
в наглядном масштабе
.
|
|
При
оптимизации эффективного заряда ядра
z
у базисных АОкоордината абсолютного минимума адиабатического потенциала равна экспериментальной длине связи.
|
|
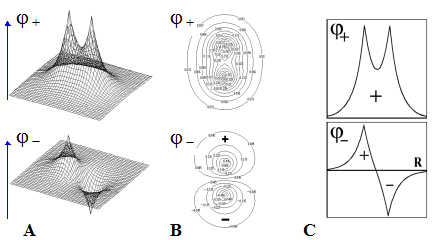 |
Признаки связывающих и разрыхляющих свойств МО
(признаки связи и разрыхления).
Рис.Графические изображения
молекулярных орбиталей
s
(
s
)- типа .
Используют три способа графического изображения МО молекулярного иона H2+ :
1) Вариант A - изображение МО в виде поверхности.
Вариант B - изображение МО в топографической форме
(в виде совокупности горизонтальных сечений - линий уровня).
3) Вариант C - изображение сечения МО вдоль линии связи.
Во всех случаях ярко выделяются пучности и узлы МО, формирующие пространственные «лепестки».
Подобное изображение возможно только для МО, базис которых составляют лишь прос-тейшие1s-АО.
|