Федеральное агентство по образованию
Федеральное государственное образовательное учреждение
высшего профессионального образования
"ЮЖНЫЙ ФЕДЕРАЛЬНЫЙ УНИВЕРСИТЕТ"
химический факультет
КУРСОВАЯ РАБОТА
Теоретическое изучение возможности изомеризации
карбенов в четырех- или шестичленные гетероциклы
Выполнил студент 4 курса
Мельниченко Д.
Научный руководитель: доцент,
кандидат химических наук
Клецкий М.Е.
Ростов – на– Дону
2008
Введение
Карбены — чрезвычайно реакционноспособные нейтральные частицы R2
C:, в которых углерод соединен с двумя группами ковалентными связями и обладает двумя несвязанными электронами [1]. Карбены являются истинными интермедиатами с характерной реакционной способностью и селективностью, которая не зависит от способа генерации, но зависит от природы заместителей R и электронного состояния частицы в момент реакции. Электронное состояние играет очень важную роль, поскольку реакции синглетных карбенов, в которых два несвязанных электрона спарены, по характеру совершенно отличаются от реакций триплетных частиц , в которых электроны с параллельными спинами расположены на разных орбиталях. Проведена большая теоретическая работа с целью предсказания электронной структуры и геометрии карбенов. Сам метилен в основном состоянии является скошенным триплетом, в котором один из несвязанных электронов находится на σ-орбитали, обладающей значительным р-характером, а другой электрон занимает р-орбиталь, перпендикулярную плоскости молекулы.

В соответствии с предсказанием первое возбужденное состояние должно быть синглетом, валентный угол в котором меньше, чем в случае триплета. Так, для метилена и многих других карбенов различия в энергии между α
-
и р
-орбиталями невелики, и поэтому триплетное состояние имеет более низкую энергию, чем синглетное состояние, вследствие большей энергии отталкиваний между электронами в последнем. В настоящее время разность в энергии между двумя состояниями оценивается в 45кДж-моль-1
. Согласно расчетам, стабилизация синглетного основного состояния за счет большего р-расщепления может быть достигнута уменьшением валентного угла или взаимодействием одной из несвязывающих орбиталей карбена с орбиталями заместителя, подающими или принимающими электроны от карбена. Примерами этого типа являются и галокарбены. Синглетное основное состояние таких карбенов, а также алкоксикарбонил и ацилкарбенов подтверждается экспериментальными данными.
Реклама

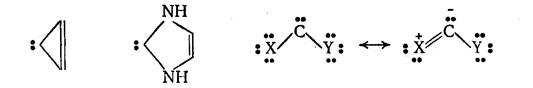
Близость синглетного и триплетного состояний и возможность их взаимных превращений представляют особый интерес в химии карбенов. Спиновое состояние карбена в момент реакции зависит не только от состояния в момент образования или от природы основного состояния, но также и от относительных скоростей процессов перехода внутри системы и реакций образования продукта.
Например, многие карбены, обычно находящиеся в триплетном основном состоянии (например, :СН2
), при регенерации в растворе образуются в синглетном состоянии и так быстро реагируют с большинством субстратов, что не успевают перейти в основное состояние.
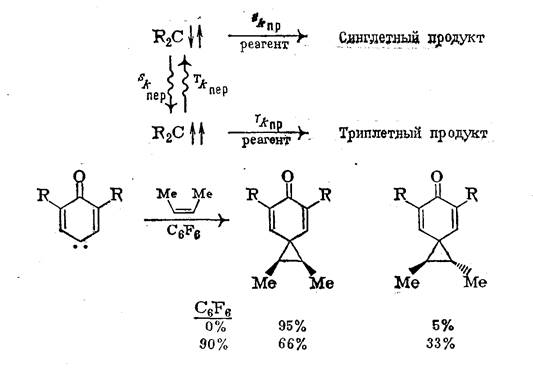
Для таких случаев разработано несколько методик, позволяющих наблюдать реакции в другом спиновом состоянии. Обычная методика основана на увеличении времени жизни карбена так, чтобы стал возможным переход между состояниями внутри системы до момента реакции. Время жизни карбена можно увеличить разбавлением реакционной смеси инертным растворителем или, что реже, генерацией карбенов с замораживанием на матрице. Например, на схеме разбавление приводит к падению стереоспецифичности реакции, что указывает на увеличение выхода продукта, за счет триплетного (основное состояние) карбена. Однако этот эффект не всегда достигается при использовании растворителей. Например, в случае присоединения дифенилметилена разбавление циклогексаном не изменяет соотношения цис/транс-продуктов.
Это обусловлено настолько близкими энергиями синглетной и триплетной частиц, что скорость установления термического равновесия сравнима со скоростью присоединения частиц к алкену.
В тех случаях, когда мультиплетность карбена нельзя определить в момент возникновения, используя разбавление, ее можно установить по изменению стереоспецифичности реакции после добавок, связывающих карбен в момент возникновения.
Другой метод увеличения переходов между состояниями до реакции основан на повышении скорости процесса путем использования растворителя с тяжелыми атомами или включением тяжелых атомов, например ртути или галогена, в самкарбен. Так, для CH3
HgCCN переходы между состояниями проходят быстрее, чем для аналогичного CH3
CCN.
Реклама

Перегруппировки карбенов
Для синглетных карбенов характерны 1,2-сдвиги. Очень легко мигрирует водород, но возможны также миграции алкильных и арильных групп, например RS и F. Эта реакция используется для синтеза алкенов против правила Бредта.
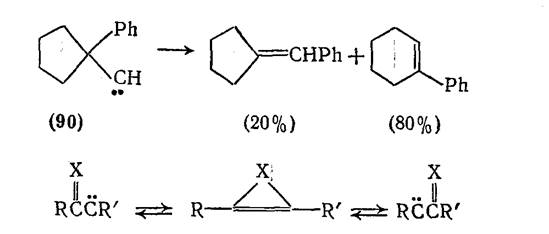
В подходящих условиях карбены, имеющие α,β-ненасыщенные заместители подвергаются карбен-карбеновым перегруппировкам. При X = О такое взаимное превращение предшествует перегруппировке Вольфа для фотолитически генерированных кетокарбенов.
К этой же группе относятся арилкарбены и, хотя обычно они не перегруппировываются в растворе, в условиях импульсного вакуумного пиролиза при температуре ниже 600°С устанавливается их равновесие с ароматическими карбенами; при более высоких температурах протекает также сужение цикла с образованием, например, винилиденциклопентадиена Внешне аналогичную серию перегруппировок претерпевают арил-нитрены.
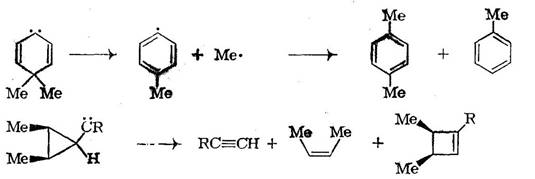
Другими характерными для карбенов перегруппировками и фрагментациями являются превращения циклопропенилидена в аллен, циклобутилидена в метиленциклопропан.
Постановка и решение задачи
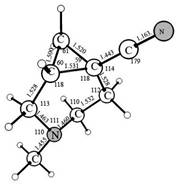
В соответствии с планом совместных работ, проводимых в рамках сотрудничества НИИ ФОХ ЮФУ и Университета Версаля (Франция) была поставлена задача изучения методами квантовой химии возможности реакций изомеризации карбена 1:
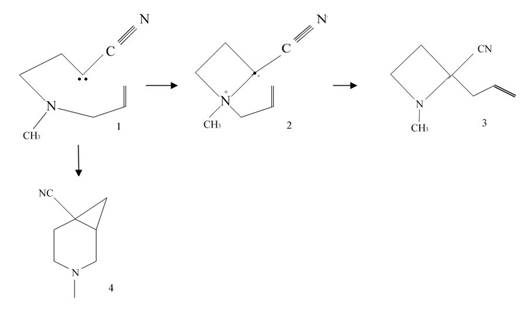
В результате таких процессов могут быть получены либо замещенные четырехчленные системы типа 3, либо бициклические системы типа 4. Все процессы экспериментально изучаются группой Ф. Кути в Университете Версаля в тетрагидрофуране (THF).
Для решения поставленной задачи нами были проведены DFT-квантовохимические расчеты в базисе B3LYP/6-31G** как в газовой фазе, так и в THF.
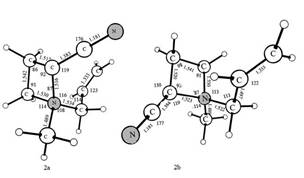
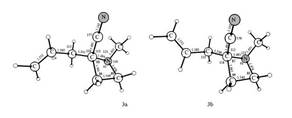
На рис. 1 представлены результаты газофазных расчетов, а на рис. 2 – расчетов в THF. Во всех случаях для систем 1-4 выбирались наиболее стабильные конформеры (соответственно 1а, 2а, 3а).
Следует отметить ожидаемую достаточную сближенность синглетного (S0
) и триплетного (T1
) электронных состояний для исходного карбена (1а,1b). Более того, по расчетам, исходный карбен 1 наиболее энергетически стабилен именно в триплетном состоянии, см. табл. 1 .
Полные и относительные энергии рассчитанных систем представлены в табл.1. Очевидно, что процессы 1→2→3 и конкурирующий с ними 1→4 протекают как сильно экзотермические (особенно последний) и, следовательно, можно утверждать, что термодинамически все рассмотренные процессы перегруппировки карбена 1 в гетероциклические продукты 3 и 4 выгодны и могут быть реализованы экспериментально.
Figure1.
Calculated geometrical attributes of isomers 1- 4 in the gas phase. Bond lengths in Ǻ, angles in degrees.

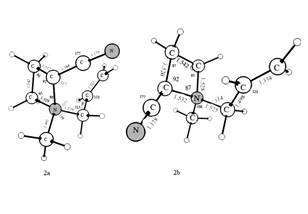
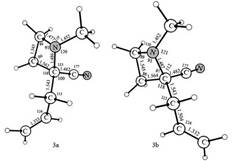
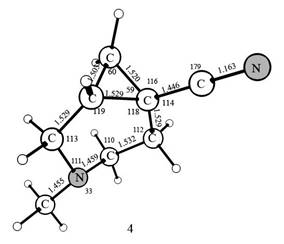
Figure 2.
Calculated geometrical attributes of isomers 1 - 4 in the THF. Bond lengths in A, angles in degrees.

 4 4
Figure3.
Schematic view of the energy gaps between the most stable isomer 1 and others in the gas phase and THF (dotted lines).

Table 1
. Calculated total (E, in a.u.) and relative (E, in kcal/mol) energies of the thermodynamically stable isomers 1-4 in the gas phase and in the THF. Values in parenthes are total and relative energies of systems 1a and 1b in triplet state.
| Gas phase
|
THF
|
| System
|
Е
|
ΔЕ
|
Е
|
ΔЕ
|
| 1
a
|
-421.41109
(-421.43179)
|
0.0
-12.9
|
-421.41730 |
0.0 |
| 1
b
|
-421.40994
(-421.42961)
|
0.7
-11.6
|
-421.41585 |
0.9 |
| 2
a
|
-421.44527 |
-21.4 |
-421.45841 |
-25.8 |
| 2
b
|
-421.44398 |
-20.6 |
-421.45667 |
-24.7 |
| 3
a
|
-421.51760 |
-66.8 |
-421.52212 |
-65.8 |
| 3
b
|
-421.51673 |
-66.3 |
-421.52234 |
-65.9 |
| 4
|
-421.53943 |
-80.5 |
-421.54595 |
-80.7 |
Методика квантовохимических расчетов
Расчеты по методу теории функционала плотности (DFT) проводились с использованием B3LYP-обменно-корреляционного функционала [2-3] и стандартного базисного набора 6-31G** [4]. Оптимизация геометрии проводилась методом аналитического расчета градиентов по схеме Берни. Природа стационарных точек устанавливалась на основании данных расчета частот нормальных колебаний (матрицы силовых постоянных).
Влияние растворителя (с полной оптимизацией геометрии всех стационарных точек) учитывалось в рамках модели поляризуемого континуума (PCM) [5-8]. В качестве растворителя был выбран тетрагидрофуран (ε = 7,58). Все расчеты выполнялись с использованием программного комплекса Gaussian 03 [9].
Литература
1. Д. Бартон, У.Д. Оллис, Общая органическая химия. Москва "Химия", 1981.
2. Becke, A.D. J. Chem. Phys. 1993, 98, 5648.
3. Lee, C., Yang, W., Parr, R. G., Phys. Rev. B 1988, 37, 785.
4. Hehre, W.J., Radom, L., Schleyer, P.v.R., Pople, J.A.. Ab initio Molecular Orbital Theory; Wiley: New York, 1986.
5. Simkin, B.Yа., Sheikhet, I.I. Quantum Chemical and Statistical Theory of Solutions: A Computational Approach; Ellis Horwood: London, 1995.
6. Cances, E., Mennucci, B., Tomasi, J. J. Chem. Phys. 1997, 107, 3032.
7. Cossi, M., Barone, V., Cammi, R., Tomasi, J. J. Chem. Phys. Lett. 1996, 255, 327.
8. Barone, V., Cossi, M., Tomasi, J. J. Comput. Chem. 1998, 19, 404.
9. Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.J.A.; Stratmann, R.E.; Burant, J.C.; Dapprich, S.; Millam, J.M.L.; Daniels, A.D.; Kudin, K.N.; Strain, M.C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G.A.; Ayala, P.Y.; Cui, Q.; Morokuma, K.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Cioslowski, J.; Ortiz, J.V.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.W.; Gill, P.M.; Johnson, B.; Chen, W.; Wong, M.W.; Andres, J.L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E.S.; Pople, J.A. Gaussian 03., Gaussian Inc.: Pittsburgh PA, 2003.
|
