Классификация каталитических процессов.
Жидкофазный металлокомплексный катализ
Все реакционные системы принято делить на гомофазные и гетерофазные. В первом случае в реакционной системе отсутствуют границы раздела фаз. Катализатор и реагенты находятся в одной фазе и в ней же протекают реакции. Гетерофазные реакционные системы имеют хотя бы одну границу раздела фаз.
Каталитические процессы классифицируют следующим образом.
1. Гетерогеннокаталитические процессы.
2. Гомогеннокаталитические процессы.
3. Процессы мицеллярного катализа.
4. Микрогетерогенный катализ.
5. Межфазный катализ.
6. Ферментативный катализ.
Гетерогеннокаталитические процессы всегда протекают в гетерофазных системах,
поскольку есть по крайней мере одна граница раздела фаз – поверхность твердого катализатора, а реагенты, продукты и другие компоненты реакционной системы могут быть газообразными и жидкими. Гомогеннокаталитические процессы могут быть как гомофазными, так и гетерофазными (гидрирование в жидкой фазе с участием растворенного в жидкой фазе катализатора – это гомогеннокаталитический процесс в гетерофазной системе, т.к. есть граница раздела фаз – Н2
/раствор, а реакция протекает в жидкой фазе, в которой растворен водород.). Реакция этерификации этанола уксусной кислотой в присутствии растворенного кислотного катализатора при отсутствии расслаивания является гомогеннокаталитическим процессом в гомофазной системе.
К катализаторам, использующимся в промышленности, есть определенные требования по следующим показателям:
1. Активность (производительность).
2. Селективность.
3. Стабильность.
4. Воспроизводимость.
5. Экономичность.
6. Экологичность.
7. Наличие методики регенерации и утилизации отработанного катализатора.
Кроме того, к гомогенным и гетерогенным катализаторам предъявляется ряд специфических требований.
Гетерогенные катализаторы – механическая прочность, определенная теплопроводность.
Гомогенные катализаторы – низкая коррозионная активность, наличие эффективного метода выделения либо катализатора из реакционной системы с последующей регенерацией и возвращением на стадию синтеза, либо продуктов реакции.
Выполнение последнего требования для эффективности гомогеннокаталитического процесса особенно важно в случае для металлокомплексных катализаторов, включающих активные переходные и в том числе благородные металлы.
Реклама

Методы разделения гомогеннокаталитических жидкофазных реакционных систем.
Существует шесть различных вариантов решения этой проблемы.
1. Выделение наиболее ценных компонентов каталитической системы (чаще всего благородных металлов) следующими приемами.
А). В газовую фазу в виде легколетучих соединений, например, карбонилов;
Б). В другую жидкую фазу (экстракция катализатора);
В). В твердую фазу (кристаллизация, восстановление).
Вариант А достаточно экзотичен и может быть реализован если продукты процесса нелетучие, а катализатор (благородный металл) находится или может быть трансформирован в легколетучие соединения.
Вариант Б возможен, если удастся подобрать экстрагент, растворитель, удовлетворяющий следующим требованиям:
- высокая селективность в отношении растворимости компонентов каталитической системы, т.е. хорошая растворимость катализатора и нерастворимость продуктов;
- нетоксичность;
- нелетучесть;
- доступность и дешевизна;
- отсутствие влияния экстрагента на характеристики каталитического процесса.
Применение варианта В может быть оправдано, если катализатор можно количественно перевести в твердую фазу за счет образования нерастворимого в контактном растворе комплекса или разложения (восстановления) растворимых комплексов до фазы компактного металла. Например, в процессе карбалкоксилирования ацетилена в эфиры насыщенных и ненасыщенных моно- и дикарбоновых кислот, катализируемом системой PdI2
-LiI-HCl в н-бутаноле палладий на 98% удается выделить за счет обработки раствора сухим аммиаком. Образующиеся аммиачные комплексы палладия практически нерастворимы в контактном растворе и могут быть отфильтрованы挹
Li2
PdI4
+ 2NH3
→ Pd(NH3
)2
I2
↓ + 2 LiI
Выделенные комплексы палладия возвращают в исходный каталитический раствор, где они под действием хлористого водорода превращаются в активные в процессе тетрагалогенидные анионы.
Pd(NH3
)2
I2
+ 4HCl → H2
PdI2
Сl2
+ 2NH4
Cl
2. Выделение продуктов гомогеннокаталитической реакции следующими приемами.
Г). Отгонка продуктов в газовую фазу (продукты легкокипящие, а каталитическая система термостабильна, не содержит легколетучих компонентов и не катализирует нежелательных превращений прдуктов и растворителя в ходе отгонки);
Д). Экстракция продуктов;
Реклама

Е). Кристаллизация продуктов.
Варианты группы 2 из общих соображений предпочтительнее вариантов группы 1 потому, что при выделении продуктов каталитическая система не разрушается, и проще организовать непрерывный процесс. Вариант Г наиболее простой и экономичный, если выполняются вышеуказанные условия.
Вариант Д связан с подбором селективного по отношению к продуктам экстрагента, удовлетворяющего требованиям, указанным для варианта Б. Сравнительно просто организовать экстракцию органического продукта, образующегося в водной каталитической системе.
Вариант Е удобно использовать для отделения продуктов, имеющих ограниченную растворимость в контактном растворе и сильную зависимость растворимости от температуры. Например, в процессе получения янтарного ангидрида из ацетилена и оксида углерода, катализируемого бромидом палладия в ацетонитриле, продукт ограниченно растворим в контактном растворе. Это позволяет выделять янтарный ангидрид за счет охлаждения контактного раствора с последующей фильтрацией без разрушения каталитической системы.
С2
Н2
+ 2СО + Н2
О → (ОССН2
СН2
СО)О (янтарный ангидрид)
Использование этих приемов будет проиллюстрировано дополнительно на примерах различных технологических схем процесса оксосинтеза.
Гидроформилирование алкенов (Оксосинтез)
Процесс оксосинтеза открыт в 1938 Роеленом. В качестве катализатора им предложен октакарбонил кобальта. Позднее установлено, что наиболее активными катализаторами гидроформилирования алкенов являются соединения родия, получившие в настоящее время широкое применение в промышленности.
Процесс гидроформилирования различных алкенов проводят в мире в промышленном масштабе - 6,6 млн. т/г (1996 г.) для получения альдегидов и спиртов. Первичными продуктами оксосинтеза являются альдегиды, которые в разной степени в зависимости от состава каталитической системы и условий превращаются в спирты.
H2
RCH=CH2
+ CO + H2
- RCH2
CH2
CHO + RCH(CHO)CH3
- RCH2
CH2
CH2
OH + RCH(CH2
OH)CH3
Механизм процесса оксосинтеза включает стадии активации и превращения водорода и алкена. В случае катализа этой реакции октакарбонилом кобальта общепринятый механизм выглядит следующим образом.
   Н2
+ Со2
(СО)8
2 НСо(СО)4 Н2
+ Со2
(СО)8
2 НСо(СО)4
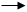 НСо(СО)4
НСо(СО)3
+ СО НСо(СО)4
НСо(СО)3
+ СО
 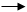 НСо(СО)3
+ RCH=CH2
НСо(СО)3
(RCH=CH2
) (π1
) НСо(СО)3
+ RCH=CH2
НСо(СО)3
(RCH=CH2
) (π1
)
 π1
+ CO RCH2
CH2
Co(CO)4
(σ1
) π1
+ CO RCH2
CH2
Co(CO)4
(σ1
)
π1
+ CO RCH(CH3
)Co(CO)4
(σ2
)
σ1
+ CO RCH2
CH2
C(O)Co(CO)4
(σ3
)
σ2
+ CO RCH(CH3
)C(O)Co(CO)4
(σ4
)
σ3
+ H2
RCH2
CH2
CHO + HCo(CO)4
σ4
+ H2
RCH(CH3
)CHO + HCo(CO)4
Активация водорода приводит к образованию гидрокарбонила кобальта. Активация алкена становится возможной после освобождения места в координационной сфере кобальта в результате диссоциации одной карбонильной группы. При взаимодействии алкена с ненасыщенным гидрокарбонилом кобальта образуется пи-комплекс, который может превратиться в два различных сигма-кобальторганических соединений в зависимости от направления нуклеофильного присоеди乱ения гидридного лиганда и кобальта с карбонильными лигандами. Далее кобальталкильные соединения подвергаются внедрению карбонильной группы из внутренней сферы комплекса с заполнением освободившегося места молекулой оксида углерода. Завершает каталитический цикл стадия гидрогенолиза с образованием альдегида линейного или разветвленного строения.
Скорость образования альдегидов описывается кинетическим уравнением
R = [alkene][Co]PH2
/(PCO
+ kPH2
)
Торможение скорости процесса парциальным давлением оксида углерода связано, по-видимому, со стадией диссоциации карбонильной группы для освобождения координационного места.
Очень важная проблема – регулирование региоселективности процесса оксосинтеза, т.е. соотношения выходов линейного и разветвленного продуктов.
В обычных условиях оксосинтеза с использованием дикобальтоктакарбонила (Co2
(CO)8
, 160-200 ат, 130-150° ) соотношение образующихся альдегидов (линейный/разветвленный) составляет ~ 4. И суть проблемы заключается в том, что потребность в разветвленных продуктах оксосинтеза (альдегидах и спиртах) существенно ниже, чем в линейных изомерах, которые необходимы для производства пластификаторов, поверхностноактивных веществ и т.д. По имеющимся данным, региоселективность оксосинтеза определяется равновесием σ-алкильных интермедиатов:
RCH2
CH2
Co(CO)3
↔
RCH(CH3
)Co(CO)3
На это равновесие влияют температура, давление синтез-газа, состав каталитической системы. Варьирование температуры не позволяет существенно повысить региоселективность в пользу линейных продуктов, т.к. повышение температуры снижает селективность, а понижение температуры приводит, кроме ограниченного повышения региоселективности, к снижению производительности процесса (рис. 2). Повышение давления также приводит к некоторому повышению соотношения образующихся продуктов в пользу линейного изомера при сохранении производительности (увеличение скорости за счет увеличения PH
2
компенсируется торможением за счет увеличения парциального давления оксида углерода (см. уравнение). К более существенному эффекту приводит введение в каталитическую систему трибутилфосфина (Bu3
P) в качестве лиганда: при этом соотношение линейный продукт/разветвленный продукт повышается до 6-7 (рис. …. ). Причиной увеличения региоселективности при использовании объемных лигандов (как и в случае повышения давления CO) считают увеличение стерических препятствий для присоединения кобальта с лигандами к внутреннему атому углерода. Это приводит к смещению равновесия σ-алкильных интермедиатов влево. Наибольший эффект в повышении доли линейных продуктов достигается при использовании родиевыз катализаторов с фосфиновыми лигандами.
Условия и показатели процесса оксосинтеза существенно зависят от природы используемого катализатора (табл. 1).
Таблица 1
Показатели процесса гидроформилирование пропилена при использовании разных катализаторов
Показатели
|
Нафтенатно-испарительные схемы |
Схема с кобальтфосфиновым катализатором |
Схема с родийфосфино-вым катализатором |
| ВНИИНЕФ-ТЕХИМ-Leuna Werke |
C рециклом олефина (ВНИИНЕФТЕХИМ) |
| Катализатор |
НСо(СО)4
|
НСо(СО)4
|
НСо(СО)3
РBu3
|
HRh(CO)(PPh3
)3
|
| Температура, о
С |
120-160 |
120-130 |
160-200 |
60-120 |
| Концентрация катализатора, %масс. |
0,1-0,5 |
0,1-0,3 |
0,6 |
0,01 |
| Степень гидрирования алкена |
Низкая |
Низкая |
Высокая |
Высокая |
| Продукты реакции |
Альдегиды и спирты |
Альдегиды |
Спирты |
Альдегиды |
| Отношение продуктов, н:изо |
80:20 |
80:20 |
88:12 |
92:8 |
Селективность процесса ,%
При расчете
на продукты нор-мального строения
на сумму целевых продуктов
|
69
87
|
76
95
|
81
90
|
78
86
|
На основе этих данных в начале 80-х считалось, что при гидроформилировании низших олефинов технологические схемы классического оксосинтеза с использованием октакарбонила дикобальта как минимум конкурентоспособны по сравнению с модифицированными схемами, в том числе с использованием родиевых катализаторов.
Однако последующее развитие событий опровергло этот вывод. Интересна динамика изменения объема производства продуктов оксосинтеза и доли процессов, использующих родиевые катализаторы (табл.2).
Таблица 2
Динамика изменения объема производства продуктов оксосинтеза и используемости катализаторов различного типа
| Год |
Объем производства, млн.т/г |
Доля продукции, полученной с использованием родиевых катализаторов, % |
| 1965 |
0,8 |
0 |
| 1980 |
5,2 |
10 |
| 1996 |
6,6 |
80 |
Приведенные данные свидетельствуют о существенной переориентации даже действующих производств на родиевые катализаторы.
Для иллюстрации вышеперечисленных методов разделения каталитических систем рассмотрим технологические схемы оксосинтеза в историческом аспекте.
Проблема разделения каталитической системы и продуктов в оксосинтезе актуальна, поскольку продукты достаточно реакционноспособны и имеют высокие температуры кипения, в кобальт в виде карбонильных соединений каталитически активен и летуч. Поэтому для процесса оксосинтеза были разработаны различные варианты выделения соединений кобальта (декобальтизации) и выделения продуктов гидроформилирования из каталитической системы.
Исторически первыми были схемы выде5ления кобальта в твердую фазу, основанные на термической декобальтизации. В этих вариантах формирование каталитически активных комплексов и их распад до металлического кобальта протекают в соответствии с уравнением (прямая и обратная стадии соответственно):
2Co + 8CO ↔ Co2
(CO)8
Направление реакции определяется давлением оксида углерода и температурой. При температуре 80-130° и давлении CO 100-150 ат равновесие реакции сдвинутов сторону образования дикобальтоктакарбонила. Если повысить температуру до 300°, а оксид углерода заменить на чистый водород под давлением 10-15 ат, то равновесие реакции сместится влево и металлический кобальт образует фазу на имеющейся поверхности или в растворе. Первый вариант термической декобальтизации был реализован в так называемой диадной схеме.
Диадная схема
Установка оксосинтеза в соответствии с диадной схемой включает два одинаковых аппарата высокого давления, заполненные неподвижной насадкой (пемзой). В одном из этих аппаратов (1 или 3, рис.) на насадку нанесен металлический кобальт. Оба аппарата снабжены коммуникациями для подвода алкена, синтез-газа и водорода. В аппарат с нанесенным на насадку кобальтом подают при нужной температуре растворитель, синтез-газ и алкен (пропилен). За счет реакции карбонилирования:
Co + CO → Co2
(CO)8
+ H2
→ HCo(CO)4
кобальт растворяется с насадки.
После определенного времени пребывания в реакторе 1 контактный раствор через фазовый сепаратор 2 поступает в реактор 3, в котором создаются условия для разложения карбонилов кобальта (РН2
10-50 ат, 250-300°). В этих условиях образовавшийся металлический кобальт садится на чистую насадку, находящуюся в реакторе 3. Освобождаемый от кобальта контактный раствор поступает на стадию гидрирования в реактор 4 (через фазовый сепаратор 2). Образующиеся бутиловые спирты разделяются в колоннах 5, а растворитель возвращается на стадию гидроформилирования. После того, как большая часть кобальта перейдет в реактор 3, функции аппаратов меняются. В реакторе 3 протекает карбонилообразование и гидроформилирование, а реактор 1 работает как декобальтизер
.
Развитием и усовершенствованием диадной схемы явилась триадная схема.
Триадная схема
Недостатком диадной схемы является периодичность работы аппаратов и образования продуктов гидроформилирования. Отсутствие непрерывности, частично сглаживаемое наличием промежуточных емкостей, осложняет работу подсистемы разделения. Для решения этой проблемы в триадной схеме появляется специализированный реактор гидроформилирования. В схему входят три основных аппарата: два аппарата выполняют поочередно функции кобальтизера (апп. 3 и 3’, схема ), в котором образуется раствор карбонилов кобальта в растворителе, и декобальтизера, а в третьем (апп. 1, схема) протекает гидроформилирование. Кобальтизер заполняется пемзой с нанесенным на нее кобальтом, а декобальтизер – чистой пемзой. По мере истощения кобальта в кобальтизере и накопления его в декобальтизере функции этих аппаратов меняются. Для быстрого перехода кобальта в карбонилы требуется давление синтез-газа ~300 ат и температура 80-300°С. Гидроформилирование проводят также при 20,0-30,0 МПа. В связи с этим все три аппарата должны быть рассчитаны на работу под высоким давлением.
Небходимость использования большого количества аппаратов под высоким давлением – основной недостаток диадной и триадной схем. Кроме этого – высокая цикличность работы отдельных аппаратов, затрудняющая организацию полностью непрерывного производства, неравномерное осаждение кобальта на насадке, невысокая производительность в расчете на единицу объема аппарата высокого давления.
Принцип термического разложения карбонилов кобальта с осаждением металлического кобальта на развитой поверхности носителя был использован в кизельгурной схеме. Только в этом варианте носитель был движущийся и это позволило в значительной мере преодолеть недостатки диадной и триадной схем.
Кизельгурная схема
Схема включает два последовательно соединенных реактора гидроформилирования (1 и 2, схема). В первый реактор катализатор поступает в виде порошка природного алюмосиликата – кизельгура, на который нанесен металлический кобальт. В первом реакторе происходит (при давлении синтез-газа 250-300 ат и температуре 150-170°С) образование (с переходом в раствор) карбонилов кобальта и частично реакция гидроформилирования. Завершается гидроформилирование во втором аппарате при температуре на 10-20° более высокой, чем в первом аппарате. Контактный раствор с суспендированным кизельгуром поступает на декобальтизацию. Декобальтизацию осуществляют в двух последовательно работающих реакторах 4 при 120-130°С и давлении водорода 2,5-3,0 МПа. Карбонильные комплексы распадаются и кобальт, осажденный на кизельгур, отделяется от раствора в магнитных сепараторах 5.
Основные недостатки этой схемы связаны, во-первых, с эрозией аппаратуры и запорной арматуры, во-вторых, со сложностью отделения суспензии катализатора от жидких продуктов с помощью магнитных сепараторов. Схема использовалась в промышленном масштабе. Кроме того, был разработан аналог схемы, в котором использовали порошок кобальта (без носителя).
Общим недостатком схем, основанных на термической декобальтизации, является частичное гидрирование альдегидов в спирты. Поэтому все вышеприведенные схемы включают узел гидрирования для получения спиртов в качестве основных продуктов. Иначе обстоит дело в случае так называемых солевых схем.
Солевые схемы
В этих схемах источником (прекурсором) карбонилов кобальта являются соли кобальта. Под действием синтез-газа (20-30 МПа) происходит восстановление кобальта с образованием карбонилов и гидрокарбонилов:
2Co2+
+ 8CO + 2H2
= Co2
(CO)8
+ 4H+
Co2
(CO)8
+ H2
=2
2 2HCo(CO)4
В реактор 1 (схема), где совмещены процессы образования карбонилов и гидроформилирования, подается соль кобальта в виде раствора или суспензии в воде или органическом растворителе, синтез-газ и алкен. При необходимых температуре и давлении происходит образование карбонилов и гидроформилирование. Продукты охлаждаются в холодильнике 2, газы отделяются в сепараторе 3. Раствор поступает в декобальтизер 4. Туда же подаются окислитель и кислота. Предложено много вариантов пары окислитель-кислота. Наиболее дешевый окислитель – кислород, в сочетании с которым могут использоваться как органические (HCOOH, CH3
COOH), так и неорганические кислоты. А фирма Мицубиси предложила использовать азотную кислоту в качестве и окислителя, и кислоты одновременно. В последнем случает в аппарате 4 протекают реакции:
3Co2
(CO)8
+ 16HNO3
= 6Co(NO3
)2
+ 4NO + 8H2
O
3HCo(CO)4
+ 7 HNO3
= 3 Co(NO3
)2
+ 3H2
+ NO + 2H2
O
Образовавшаяся соль кобальта либо растворяется в водном слое, либо образует суспензию, и может быть отделена от органических продуктов и возвращена в реактор 1.
Несколько отличный вариант солевлй схемы известен под названием кульмановского
процесса. Активным катализатором в этом случае также является гидрокарбонильный комплекс кобальта – Hco(CO)4
. Процесс протекает при 110-180°С и давлении синтез-газа 20-25 МПА в реакторе 1 (схема) с интенсивными массообменом газ-жидкость и теплообменом. В течение 1-2 ч достигается степень превращения алкена 90-95% и селективность образования альдегидов 85-90%. Из реактора 1 поток, содержащий продукты реакции, попадает в колонну 2, где катализатор HCo(CO)4
экстрагируется из органической фазы водным раствором Na2
CO3
; при этом протекает реакция, в ходе которой кобальт восстанавливается, а восстановителем является сам гидридный лиганд:
2HCo(CO)4
+ Na2
CO3
= 2NaCo(CO)4
+ CO2
+ H2
O
В фазовом сепараторе 3 удаляются остаточные газы. Оставшийся катализатор экстрагируется водой из органической фазы во второй поглотительной колонне 2. Водные растворы из двух поглотительных колонн подают в колонну 4, в которой их обрабатывают кислотой. При этом кобальт окисляется протоном с образованием активной формы катализатора:
2NaCo(CO)4
+ H2
SO4
→ 2HCo(CO)4
+ Na2
SO4
Гидрокарбонил кобальта, образующийся в этой реакции, летуч и умереннорастворим в водной кислоте. Он выносится в верхнюю часть колонны током синтез-газа, подаваемого снизу, и поступает в нижнюю часть адсорбционной колонны 5. В верхнюю часть той же колонны 5 подается алкен, разбавленный растворителем. Образующийся в колонне 5 раствор регенерированного катализатора, содержащий алкен, направляют в реактор 1. Продукты, поступающие из второй поглотительной колонны 2, подвергают дальнейшим стадиям очистки.
Во всех вариантах схем реакции карбонилообразования и гидроформилирования идут при высоком давлении (20-30 МПа), а стадия декобальтизации, проводимая чаще всего за счет экстракции соли кобальта в водную фазу, осуществляется при низком (практически атмосферном) давлении.
Основными достоинствами солевых схем оксосинтеза следует считать уменьшение объема оборудования высокого давления, полная непрерывность работы установки, уменьшение потерь альдегидов за счет гидрирования в спирты.
Недостатки – относительное усложнение стадиии декобальтизации, введение дополнительных реагентов, появление сточных вод и коррозия оборудования. Этих недостатков в основном лишены испарительные схемы.
Испарительные и смешанные схемы
Наиболее простым с технологической точки зрения вариантом отделения продуктов гидроформилирования от каталитической системы является их отгонка. Для реализации этого варианта необходимо выполнение двух условий. Прдукты гидроформилирования должны быть относительно низкокипящими, а катализатор должен быть переведен в нелетучее и неактивное состояние. Для кобальтовых катализаторов наилучший вариант - нафтенатно-испарительная схема.
Нафтенантно-испарительная схема
Эту схему широко использовали и используют для получения низших альдегидов (пропионовый, масляные, амиловые). Кобальтовый катализатор перед отгонкой превращают в кобальтовые соли нафтеновых кислот - нелетучие и каталитически неактивные соединения.
Раствор нафтеновых солей кобальта (свежий и регенерированный) в соответствующем растворителе поступает в реактор карбонилообразования 1 (схема). Туда же подают синтез-газ, переводящий катализатор в активную форму:
2Co(RCOO)2
+ 8CO + 3H2
→ 2HCo(CO)4
+ 4RCOOH
Каталитически активный раствор поступает в реактор гидроформилирования 2, в который подают дополнительное количество синтез-газа и алкен. Контактный раствор после определенного времени пребывания в реакторе 2 через фазовый генератор поступает в аппарат 4, в котором происходит окисление кобальта, как правило, кислородсодержащим газом, с образованием солей нафтеновых кислот:
2HCo(CO)4
+ 4RCOOH + 1.5O2
→ 2Co(RCOO)2
+ 3H2
O
Раствор продуктов гидроформилирования с нафткнатом кобальта направляют через фазовый сепаратор 3 в ректификационную колонну 5. В колонне 5 отгоняют продукты гидроформилирования (альдегиды), а кубовый остаток, содержащий растворитель, побочные продукты и нафтенат кобальта, возвращают в реактор 1. Потери кобальта при использовании этого варианта не превышают, как правило, 1%.
Наиболее эффективными испарительные схемы оказались при использовании родиевых катализаторов с фосфиновыми лигандами.
|
