Министерство образования и науки Украины
Донецкий национальный университет
Химический факультет
Кафедра физической химии
ДИПЛОМНАЯ РАБОТА
На тему: «Анализ сополимеризации индена с малеиновым ангидридом»
Специальность: 7.0703301 – ф/х «Химия»
Дипломник:
Мелентьев Иван Сергеевич
Руководитель:
д.х.н., проф. Заварин Дмитрий Петрович
Донецк – 2009
Список условных обозначений
n –количество измерений
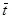 -среднее время истечения -среднее время истечения
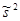 - исправленная выборочная дисперсия - исправленная выборочная дисперсия
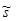 - выборочное среднеквадратическое или стандартное отклонение - выборочное среднеквадратическое или стандартное отклонение
ε – доверительный интервал
Р –доверительная вероятность
МА – малеиновый анегидрид
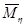 - средневязкостная молекулярная масса - средневязкостная молекулярная масса
СПЛ – сополимер
КО - комплексообразователь
[η] – характеристическая вязкость
МА – малеиновый анегидрид
КИФ – кумарон-инденовая фракция
ИФ – инденовая фракция
ПБ - пероксид бензоила
Введение
Первоначально интерес к инден-кумароновым смолам был вызван тем, что они являлись альтернативой более дорогим синтетическим полимерам, поскольку сырьевой базой для них служат продукты переработки каменного угля. Благодаря достаточно высоким пластифицирующим свойствам, хорошей химической стойкости и водостойкости, а также относительной дешевизне эти смолы начали успешно применяться в лакокрасочной промышленности. Было показано, что они обладают и другими весьма ценными свойствами: высокой связующей и клеющей способностью, малой электро- и теплопроводностью, неплохой термостойкостью и способностью совмещаться с высыхающими маслами. Основным сырьем для их получения остаются продукты коксохимии, хотя возможно также использование побочных продуктов пиролиза нефти. Наличие такой сырьевой базы, хорошие технологические и эксплуатационные свойства при многообразии направлений использования инден-кумароновых смол способствуют сохранению устойчивого интереса к этому виду синтетических смол. В Донбассе проблема получения этих ценных смол с использованием кумарон-инденовой фракции (КИФ), многотоннажного отхода коксохомического производства, также достаточно актуальна.
Сополимеризация содержащихся в составе КИФ непредельных соединений в присутствии малеинового ангидрида и пероксидного инициатора может способствовать понижению температуры процесса получения смол, расширяет возможности использования продуктов на их основе, хотя этот вопрос изучен пока недостаточно.
Реклама

Целью данной работы является изучение полимеризации инденовой фракции в присутствии МА и изучение молекулярной массы и полученных полимеров.
1. Обзор литературы
1.1 Теоретические основы процесса комплексно-радикальной полимеризации
Полимеризация виниловых и диеновых соединений представляет собой особый вид цепной реакции, характерной особенностью которой является то, что развитие кинетических цепей сопровождается ростом молекулярных цепей из молекул мономера [1, 2]. Для цепной полимеризации [1, 2] характерно очень быстрое присоединение молекул мономера друг к другу без выделения побочных продуктов. Все способы инициирования полимеризации можно разделить на два класса. В одних случаях инициирование представляет собой реакцию присоединения к двойной связи мономера свободного радикала R*, образовавшегося тем или иным путём, а в других оно осуществляется в результате взаимодействия молекулы мономера с молекулами веществ, являющихся кислотами или основаниями Льюиса.
1.1.1 Общие положения радикальной (со)полимеризации
Процесс радикальной полимеризации можно изобразить следующей схемой [1]:
| 1. |
Реакция инициирования, приводящая к образованию из мономерных молекул М1 реакционных центров 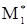 |
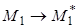 |
| 2. |
Развитие реакционной цепи через активные полимеры, сопровождающееся ростом молекулярной цепи. Активные полимеры – промежуточные продукты полимеризации. |
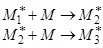 |
| 3. |
Обрыв реакционной цепи, приводящий к образованию конечного продукта – неактивного полимера Рn. |
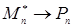 |
Рассмотрим подробнее процесс радикального инициирования. Инициаторы [2] представляют собой термически неустойчивые соединения, распадающиеся с образованием свободных радикалов. Свободный радикал R* образуется вследствие гомолитического распада молекулы инициатора при поглощении ею энергии: R:R → R·. Он атакует двойную связь в молекуле мономера, при этом свободно-радикальный активный центр перемещается с фрагмента инициатора на мономерное звено:
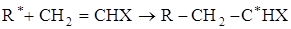 (2.1) (2.1)
Этот процесс электронной перестройки сопровождается высвобождением энергии порядка 20 ккал (80 кДж), так как p-электронный уровень расположен выше уровня s-электронов. Таким образом, свободно-радикальная атака мономера при инициировании полимеризации - экзотермический процесс, в то время как разложение инициатора на свободные радикалы – эндотермическая реакция. Разложение инициаторов на свободные радикалы может происходить под действием тепла, света или других видов энергии, а также под влиянием катализаторов. В качестве инициаторов в основном используют азосоединения, пероксиды, гидропероксиды, перэфиры и перкислоты. Скорость разложения инициаторов зависит от их химического строения, а также от температуры реакции и используемого растворителя.
Реклама

Одним из наиболее употребляемых инициаторов виниловой полимеризации является пероксид бензоила [1]. В настоящее время можно считать установленным, что при нагревании растворов пероксида бензоила во многих растворителях первичным процессом является распад пероксида бензоила на два бензоатных радикала:
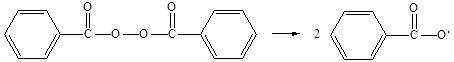 (2.2) (2.2)
которые в дальнейшем способны распадаться с выделением СО2 и с образованием фенильных радикалов:
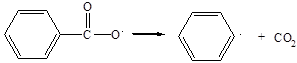 (2.3) (2.3)
Если распад пероксида производится в присутствии энергичных акцепторов радикалов, то реакция (2.3) подавляется. Снижение выхода СО2 в присутствии виниловых соединений наблюдали японские авторы [3]. В то же время при распаде пероксида бензоила в четырёххлористом углероде происходит выделение СО2 в количестве, соответствующем 96 % от теоретически возможного.[4] Так как бензоатный радикал, очевидно, не реагирует с четырёххлористым углеродом, то в этом случае почти все бензоатные радикалы распадаются согласно реакции (2.3). Кинетика распада пероксида бензоила в различных условиях была подробно исследована [3-9]. Полученные при этом результаты в основном сводятся к следующему.
1. Скорость распада пероксида бензоила сильно зависит от растворителя, в котором протекает реакция.
2. Кинетический порядок реакции распада также зависит от растворителя.
3. При уменьшении концентрации пероксида удельная скорость распада (скорость, отнесённая к начальной концентрации пероксида) во многих случаях уменьшается и для различных растворителей стремится к одинаковой величине.
4. Добавление некоторых веществ, в частности виниловых соединений, к растворителям, в которых протекает быстрый распад пероксида, приводит к снижению удельной скорости реакции до величины, наблюдаемой при распаде пероксида в разведенных растворах.
Таким образом, распад пероксида представляет цепную реакцию [6-8], причём длина цепей зависит от природы растворителя. Предложено [6] следующее выражение для скорости распада пероксида бензоила:
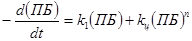 , (2.4) , (2.4)
где (ПБ)-концентрация пероксида бензоила, k1 –константа скорости первичного мономолекулярного распада, а член kц(ПБ)n характеризует скорость цепного распада пероксида.
При виниловой полимеризации скорость и эффективность инициирования определяется первичным распадом пероксида, поэтому важно знать константу скорости мономолекулярного распада пероксида kПБ. Данные распада пероксида бензоила в бензоле (начальная концентрация 0,00185 моль/л) при температуре 60-80ºС удовлетворяют уравнению [5]:
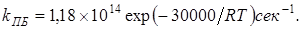 (2.5) (2.5)
В ароматических растворителях, например в толуоле, цепной распад пероксида бензоила при концентрациях пероксида не больше 0,2 моль/л невелик и приводит к образованию несимметричных дифенилов и значительных количеств бензойной кислоты (~50% от теории). Лёгкость присоединения фенильного радикала к бензольному кольцу с образованием нереакционного радикала позволяет понять малую величину цепного распада пероксида в ароматических растворителях. По-видимому, бензоатные радикалы также могут присоединяться к ароматическому кольцу, что приводит к образованию эфира бензойной кислоты. Присоединение бензоатного радикала к бензолу протекает медленнее, чем декарбоксилирование, так как выход эфира составляет лишь 5-7% [9].
Цепной распад пероксида может быть подавлен добавками ингибиторов. Особенно эффективны в этом отношении виниловые соединения. Поведение виниловых соединений по отношению к пероксиду бензоила во многих отношениях аналогично поведению ароматических соединений. Бензоатные радикалы, первоначально образующиеся при распаде пероксида бензоила, могут или присоединяться к двойной связи, давая начало полимерным цепям, или отщеплять молекулу диоксида углерода с образованием фенильного радикала, который также может присоединяться к двойной связи. Конкуренцию между реакцией декарбоксилирования и реакцией присоединения к двойной связи исследовали по выходу диоксида углерода при распаде пероксида бензоила в присутствии мономера. При увеличении концентрации мономера выход СО2 уменьшается, а при равных концентрациях зависит от природы мономера.
За инициированием следует стадия роста цепи [1, 2]. На этой стадии активный центр, находящийся на первом мономерном звене, атакует двойную связь следующей молекулы мономера. Эта атака приводит к присоединению второго мономерного звена и переносу активного центра с первого на второе мономерное звено в соответствии со следующей схемой:
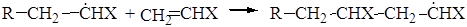
Необходимо отметить, что этот активный центр вновь способен к атаке следующей молекулы мономера с дальнейшим переносом неспаренного электрона на конец растущей цепи. Такой процесс, включающий последовательность актов присоединения молекул мономера к активному центру, получил название роста цепи. Возможны различные типы присоединения молекул мономера к активному концу растущей цепи. Так, различают присоединение по типу «голова к хвосту», «хвост к хвосту», «голова к голове», «хвост к голове», при этом под «головой» и «хвостом» мономерного звена понимают группы –СНХ– и –СН2–. Как упоминалось выше, присоединение очередного мономерного звена к концу растущей цепи сопровождается переходом p-электронной пары на уровень s-электронов и выделением энергии ~ 20 ккал. Следовательно, необходимо небольшое количество энергии извне для разложения инициатора и образования свободных радикалов, а далее полимерные цепи начинают рост с выделением большого количества энергии.
Поскольку при разложении инициатора одновременно образуется большое количество свободных радикалов, каждый из которых инициирует и продолжает рост цепей, в системе в любой момент времени существует определённое количество растущих цепей. В зависимости от ряда факторов, таких как температура, время реакции, концентрация мономера и инициатора, существует некоторая статистическая вероятность сближения двух растущих цепей и их взаимного столкновения. Когда происходит такое взаимное проникновение растущих клубков, возможно осуществление двух реакций, приводящих к обрыву цепей:

В первом случае две растущие цепи соединяются за счёт спаривания одиночных электронов активных центров, поэтому такой вид обрыва получил название рекомбинации. Во втором случае атом водорода отрывается от конца одной из растущих цепей, присоединяется к концу второй и образует стабильную связь с неспаренным электроном активного центра. Цепь, отдавшая атом водорода, стабилизируется за счёт образования концевой двойной связи. Такая реакция обрыва приводит к образованию двух полимерных молекул и носит название диспропорционирования.
Существует ещё один вид реакций ограничения длины растущих цепей, который происходит путём «передачи цепи» [2]. Эта реакция протекает обычно путём отрыва атома водорода или любого другого подвижного атома от молекулы инициатора, мономера, мёртвых полимерных цепей или любых молекул, присутствующих в реакционных системах, включая растворитель и примеси. Ее можно схематически представить следующим образом:
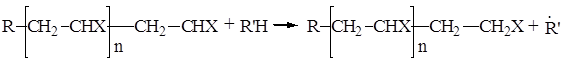
Растущая цепь при этом обрывается, но образуется новый радикал 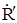 . Он теперь способен инициировать рост новой полимерной цепи. Таким образом, в ходе этой реакции одна живая, растущая цепь прекращает свой рост, тогда как новая начинает всё сначала. . Он теперь способен инициировать рост новой полимерной цепи. Таким образом, в ходе этой реакции одна живая, растущая цепь прекращает свой рост, тогда как новая начинает всё сначала.
1.1.2 Полимеризация виниловых мономеров в присутствии комплексообразователей типа кислот Льюиса
Известно, что органические основания Льюиса (нитрилы, сложные эфиры, амиды и т. п.) способны образовывать достаточно прочные координационные комплексы с координационно-ненасыщенными соединениями непереходных металлов (чаще всего Zn, B, Al, Sn и др.), выступающих в таких случаях в качестве Льюисовых кислот. Непредельные органические соединения указанных выше классов, в частности, практически важные акриловые и метакриловые мономеры обычно полимеризуются по радикальному механизму. Образование их комплексов с кислотами Льюиса, естественно, приводит к изменению электронной структуры молекул мономеров и, следовательно, их реакционной способности. Поэтому кислоты Льюиса могут быть использованы в качестве модификаторов для целенаправленного изменения кинетических параметров отдельных элементарных стадий полимеризационных процессов.
В зависимости от природы мономеров и растворителя кислоты Льюиса могут образовывать донорно-акцепторые комплексы нескольких типов. В основе существующей классификации лежит тип орбиталей, участвующих в образовании межмолекулярных связей [10]. Соединения - доноры электронов подразделяются на три группы: n, s и p в зависимости от типа высшей из занятых орбиталей (неподелённая электронная пара, s-связь и пара p-электронов соответственно). Координационно-ненасыщенные соединения металлов участвуют в комплексах за счёт вакантных орбиталей (V) атома металла. Наибольший интерес представляют следующие комплексы мономеров с кислотами Льюиса: nV-комплекс, в котором в качестве донора электронов выступает гетероатом с неподелённой парой заместителя мономера или радикала роста, и nVp-тройной комплекс, в котором акцептором является двойной комплекс nV, а донором – мономер или растворитель электронодонорного характера. Хотя те и другие комплексы по своей природе являются донорно-акцепторыми (ДА), в литературе это название в основном применяется к комплексам второго типа, а nV-комплексы по сложившейся терминологии называются координационными.
Комплексообразование между мономерами и кислотами Льюиса в координационных nV-комплексах надёжно установлено по данным ИК-, УФ- и ЯМР-спектроскопии. В ИК спектрах комплексов метилметакрилата и (мет)акрилонитрила с галогенидами металлов наблюдаются существенные сдвиги полос поглощения [11,12], соответствующих валентным колебаниям nС=О и nС≡N величиной от 20 до 155 см-1 и от 25 до 50 см-1. Частота валентных колебаний С=С связей изменяется незначительно, от 2 до 5 см-1. На основе этих данных сделан вывод о том, что комплексообразование указанных мономеров идёт преимущественно по функциональным группам заместителей и что роль двойной связи в этом процессе незначительна. Об этом свидетельствует и тот факт, что сдвиги полосы nС=О ММА и его предельного аналога – этилацетата при их взаимодействии с кислотами Льюиса близки. Весьма полезная информация получена также при анализе изменений частот деформационных колебаний δСН (в пределах от -12 до +22ºсм-1) и скелетных колебаний пиридинового кольца (21-30ºсм-1) в комплексах винилпиридинов с галогенидами цинка [13]. Природа выявленных эффектов была интерпретирована в предположении, что ион металла комплексообразователя участвует в системе сопряжения мономера и смещает на себя его электронную плотность.
Для оценки изменения степени сопряжения двойной связи акриловых мономеров при комплексообразовании было использовано смещение полосы поглощения 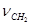 неплоских веерных колебаний С-Н связей метиленового фрагмента >C=СН2. Согласно [14], увеличение сопряжения двойной связи мономера приводит к повышению частоты , а усиление полярности - к её уменьшению. Следовательно, экспериментально наблюдаемый факт увеличения на 12-17ºсм-1 частоты колебаний (мет)акриловых мономеров (бутилакрилата, акрилонитрила, метилметакрилата) в комплексах с с хлоридами цинка и олова свидетельствует об увеличении степени сопряжения двойной связи мономера. неплоских веерных колебаний С-Н связей метиленового фрагмента >C=СН2. Согласно [14], увеличение сопряжения двойной связи мономера приводит к повышению частоты , а усиление полярности - к её уменьшению. Следовательно, экспериментально наблюдаемый факт увеличения на 12-17ºсм-1 частоты колебаний (мет)акриловых мономеров (бутилакрилата, акрилонитрила, метилметакрилата) в комплексах с с хлоридами цинка и олова свидетельствует об увеличении степени сопряжения двойной связи мономера.
Влияние комплексообразования на степень сопряжения двойной связи было доказано также методом УФ-спектроскопии [10]. Сопряжение в мономере косвенно может быть оценено по значению параметра Q схемы Q – e Алфрея – Прайса. Была установлена [13] линейная кореляция между lg Q и λπ-π*- полосой поглощения двойной связи мономера в УФ-спектре. Тазуке и Окамура [13] нашли, что при образовании комплекса с кислотами Льюиса наблюдается красное смещение полосы поглощения двойной связи. Этот факт указывает на увеличение резонансной стабилизации мономера в комплексе за счёт дополнительного сопряжения π-электронной системы мономера с атомом металла кислоты Льюиса. Уменьшение частоты поглощения, вызванное комплексообразованием, в некоторых случаях достигает 700 – 1500 см-1, что отвечает возрастанию параметра Q комплексо-связанных мономеров в 1,5 – 2 раза [15].
Об изменении полярности двойной связи мономеров при комплексообразовании свидетельствуют также данные ПМР спектров комплексов акриловых мономеров, например ММА, с хлоридами олова, алюминия и BF3, в которых химические сдвиги протонов мономеров смещены в область слабого поля по сравнению со свободными мономерами на 0,02-0,60 м.д. [11].
2 Полимеризация индена и кумарона
Интерес к полимеризации индена и кумарона был первоначально вызван тем, что они являются основными ненасыщенными компонентами сырого бензола и каменноугольного масла, способными к смолообразованию. До настоящего времени сырьем для производства инден-кумароновых смол остается тяжелый бензол, называемый также по преобладающим компонентам инден-кумароновой фракцией, хотя большую ценность представляет также и ксилольно-тяжелая фракция коксо-химического производства. Эти фракции содержат 50-70 и 35-40 % непредельных соединений и обе пригодны для получения кондиционных инден-кумароновых смол. Следует отметить, что в обеих указанных фракциях третьим по объему содержания ненасыщенным компонентом является стирол.
Наибольший интерес из указанных выше продуктов коксо-химического производства представляет инден, концентрация которого выше, чем стирола и кумарона. Инден легко бромируется, алкилируется, цианэтилируется. Однако основной областью его использования является процесс получения инден-кумароновых смол. Протекание этого процесса определяется ходом полимеризации и сополимеризации индивидуальных смолообразующих компонентов сырья. Не случайно поэтому большое число исследований было посвящено изучению химизма и условий полимеризации соединений инденового и кумаронового ряда. Начало им было положено работами Кремера и Шпилькера, опубликованными в 1890 г. [14, 16], а затем в разное время этот вопрос изучали Штермер, Штоббе и Фербер, Уитби и Кац, Штаудингер [14, 16], в 60-е годы ХХ в. обстоятельные исследования были выполнены Марешалем и др [15,16,17,18]. Уже в первых работах было установлено, что полимеризация индена может протекать под действием света, тепла, давления и главным образом под влиянием различных катализаторов.
2.1 Основные закономерности полимеризации индена и кумарона
Полимеризация индена протекает под действием солнечного света, однако чрезвычайно медленно: при выдержке на свету в течение года удалось получить лишь небольшой выход полимера, содержащего от 8 до 16 молекул исходного мономера. Довольно подробно изучена, например, фотодимеризация индена [16, 19]. Показано, что реакция протекает как через синглетное, так и через триплетное состояние индена. При несенсибилизированной фотодимеризации 80 % димера образуется через триплетный инден.
Более интенсивно протекает термическая полимеризация. Уже при обычной температуре происходит автополимеризация индена, скорость которой быстро возрастает с повышением температуры. Однако даже при температуре ~200ºС для обеспечения полноты реакции полимеризации требуется длительное время [14, 16]. Наряду с полимеризацией при нагревании индена протекает реакция автовосстановления, в результате которой происходит образование трускена (С27Н18) и гидриндена (С9Н10), а если нагревать инден в присутствии воздуха, то происходит также его автоокисление. При повышении температуры молекулярная масса продукта уменьшается, например, от 886 до 676 при 178ºС и 200ºС. Как правило, молекулярная масса продукта реакции при термополимеризации не превышает 1000.
Термополимеризация кумарона и индена во многом сходны, отличие заключается в степени и скорости полимеризации. Если, например, при 20 ч нагревания индена с обратным холодильником было получено 30% смолы, то кумарон при нагревании в тех же условиях образовал лишь незначительное ее количество [14, 16]. Молекулярная масса при термической полимеризации кумарона также ниже, чем при полимеризации индена.
Полимеризация индена и кумарона может быть инициирована различными способами. Радикальная полимеризация этих соединений изучена мало. Описана радикальная полимеризация и сополимеризация индена и кумарона в массе при давлении до 10000 атм в присутствии пероксида бензоила [17, 19]. Инден полимеризуется в течение нескольких минут; при достижении 175ºС реакция носит взрывной характер. В результате реакции образуется твёрдый жёлтый полимер. Полимеризация кумарона протекает несколько медленнее, взрывной характер реакция приобретает только при 275ºС.
Наряду с ростом макромолекулы образовавшиеся свободные радикалы способствуют протеканию и других реакций. Например, бензильный радикал отрывает Н из положения 1 у индена с образованием толуола и нового радикала, который превращается далее в 1-бензилинден и 1,1-диинденил. Возможно также гомолитическое замещение в положениях 2 и 3 у индена, сопровождающееся присоединением PhCH2 по двойной связи и образованием 2- и 3-бензилинденов [20].
Радикальная полимеризация компонентов коксохимического сырья протекает с различной скоростью. Так, под действием 5 % гидроперекиси изопропилбензола при температуре 125ºС инден полимеризуется на 46-50 %, стирол и метилстиролы – на 89-90 %, кумарон практически совсем не полимеризуется [21].
Исследована, также электроинициированная полимеризация индена. При пропускании электрического тока через раствор перхлората лития в уксусном ангидриде, содержащий инден, в анодном пространстве происходит образование полииндена. Предполагается, что реакция развивается по ионному механизму, причем максимальный выход полимера достигает 16 %. Образующийся полиинден по структуре не отличается от полимера, полученного катионной полимеризацией.
Каталитической полимерации индена и кумарона посвящено наибольшее количество работ. Еще более века назад было установлено, что эти соединения способны полимеризоваться под действием различных катализаторов кислотного типа, например, серной кислоты и AlCl3. Ранние исследования были посвящены главным образом сернокислотной полимеризации. Под действием 75 %-ной кислоты образовывалась растворимая в бензоле смола, из которой удалось выделить диинден и тетрамер индена, названный параинденом, с температурой плавления 210ºС. При действии 95 %-ной серной кислоты были получены продукты, содержащие 16-22 молекулы индена с температурой плавления 220-280ºС (метаинден).
Кумарон под действием серной кислоты также дает продукты различной степени полимеризации, среди которых охарактеризованы α- и β-паракумарон со степенью полимеризации 4 и 8 соответственно.
Кроме серной кислоты, полимеризация указанных соединений может эффективно протекать под действием таких катализаторов, как ZnCl2, FeCl3, SnCl4, TiCl4, SbCl5, AlBr3, AlCl3, BF3. Во всех этих случаях происходит так называемая гомогенная (иногда псевдогомогенная) полимеризация, при которой катализаторы вводятся в реакционную среду либо в виде раствора, либо в виде жидких комплексов солей с растворителем. Возможно также применение гетерогенных катализаторов, таких как природные или синтетические алюмосиликаты, активированные минеральными кислотами, а также оксидами или солями [22].
Согласно современным представлениям для начала катионной полимеризации необходимо возникновение активных центров. В их образовании принимают участие катализатор – галогенид металла МХn, являющийся кислотой Льюиса, сокатализатор ВН (кислота Бренстеда, чаще всего, вода иди галоидводороды), выступающий в качестве донора протона, и исходный мономер. Как и при радикальном инициировании, процесс катионной полимеризации протекает в несколько стадий: инициирование, рост цепи и его прекращение, которое осуществляется в результате передачи цепи за счет переноса протона на другую частицу: молекулу мономера, растворителя, противоион или макромолекулу.
В целом при полимеризации индена наблюдаются общие закономерности, характерные для катионной полимеризации других непредельных соединений, в частности, виниловых мономеров. Течение процесса полимеризации и его результаты очень сильно зависят от природы применяемого катализатора. Для индена наибольшая характеристическая вязкость получена в присутствии BF3 и TiCl4. Степень полимеризации с увеличением концентрации катализатора до некоторого предела растёт, после чего остаётся почти постоянной.
Кроме того, при полимеризации индена отмечена характерная для катионной полимеризации зависимость скорости и степени полимеризации от природы растворителя, а именно повышение степени полимеризации с увеличением диэлектрической проницаемости растворителя. Высокую степень полимеризации обеспечивает применение в качестве растворителей галоидированных алифатических углеводородов, особенно хлористого метилена. При использовании ароматических растворителей, например толуола, степень полимеризации существенно понижается, что может быть связано с явлением передачей цепи, которая более ярко выражена в случае ароматических соединений.
Катионная полимеризация индена возможна в довольно широком диапазоне температур, от 273 до 173 К. При этом характеристическая вязкость увеличивается от 0,53 до 1,90 дл/г (растворитель – хлористый метилен, 100 г/л индена, катализатор - TiCl4 в концентрации 0,02 моль/л)[23]. В этих же условиях при температуре 201 К молекулярная масса полииндена со снижением концентрации мономера до 50 г/л возрастает, а при дальнейшем уменьшении его концентрации также уменьшается. Это может быть объяснено, с одной стороны, снижением температуры реакции при меньших концентрациях мономера, с другой стороны, увеличением диэлектрической проницаемости среды за счёт повышения концентрации растворителя.
При благоприятных условиях полимеризации – низкой температуре реакции и применении растворителя с высокой диэлектрической проницаемостью - может быть получен полиинден с характеристической вязкостью около 2 дл/г, что соответствует молекулярной массе более миллиона [16].
Изучение катионной полимеризации кумарона в различных условиях [24] по методике, описанной ранее для индена, показало, что слабые катализаторы катионного типа не инициируют полимеризацию кумарона. Наиболее активным катализатором, как для индена, является BF3. Полимеризация кумарона с ним в растворителе с высокой диэлектрической проницаемостью при 201 К заканчивается за 5 мин и приводит к получению полимера с характеристической вязкостью 3,8 дл/г, что соответствует степени полимеризации не менее 8500. Как и для индена, полимеризация кумарона зависит от природы и диэлектрической проницаемости растворителя. Молекулярная масса полимера также увеличивается с понижением температуры реакции. В целом можно отметить, что полимеризация кумарона при равных условиях протекает медленнее, чем полимеризация индена, и с меньшим выходом, но полимер имеет более высокую характеристическую вязкость. Установлено, что по скорости катионной гомополимеризации с большинством катализаторов основные непредельные компоненты в составе коксохимического сырья располагаются в ряд : инден > стирол » кумарон [25].
2.2 Структура продуктов полимеризации индена и кумарона
Вопрос о структуре образующихся полимеров с самого начала исследований был предметом длительных научных споров. Димеру индена была приписана следующая насыщенная структура:
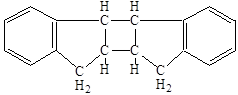 , ,
подтвержденная более поздними исследованиями [18].
Аналогично в виде замкнутых насыщенных структур представляли первоначально молекулы тетрамера и более высокомолекулярных полимеров индена. Однако такое представление противоречило экспериментальным данным о наличии остаточной непредельности у продуктов полимеризации индена, поэтому для изображения дииндена предложили формулы:
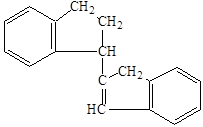 или или 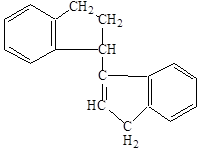 , ,
которые были перенесены на строение продуктов более глубокой степени полимеризации. Такая структура полимеров объясняет их способность присоединять галоид (наличие двойной связи в молекуле полимера), а также способность продуктов низкой степени полимеризации к дальнейшему уплотнению, и наблюдаемую даже у твёрдых хрупких смол способность к присоединению кислорода с образованием пероксидов. Этим же объясняется постепенное уменьшение иодного числа у полимеров большей молекулярной массы, так как остаточная двойная связь приходится на всё более длинную молекулу полимера.
Спектроскопические исследования [26 - 28] кумарона и индена и полученных из них методом каталитической полимеризации продуктов подтверждают, что реакция полимеризации протекает за счёт раскрытия двойной связи пятичленного цикла. Поэтому представление о линейной структуре полимеров индена и кумарона получило наибольшее распространение. Длительное время полимеры кумарона и индена представляли в виде линейных цепей с расположением молекул по одну сторону от главной оси. Экхардт и Хайне [26] впервые высказали предположение о том, что под действием катализаторов катионной полимеризации, таких, как хлористый алюминий или трёхфтористый бор, из индена возможно образование трёх типов стереорегулярных полимеров: изотактических, атактических и синдиотактических, что доказывают и исследования ИК спектров полииндена [27].
Вполне определённо доказана возможность получения стереорегулярных полимеров из кумарона [23]. Характерной особенностью кумарона является то, что он склонен к образованию оптически активных полимеров, имеющих диизотактическую структуру. В ИК спектре продукта термической полимеризации кумарона обнаружена полоса при n=3540 см-1 [27], которая может быть объяснена только образованием в поликумароне внутримолекулярной водородной связи между кислородом фуранового цикла связанной молекулы и одним из водородов другой молекулы, которая может возникнуть только в том случае, если цепь поликумарона имеет не линейное, а зигзагообразное строение:

Регулярное расположение молекул в цепи поликумарона должно быть устойчивым, так как оно стабилизируется водородной связью. Получение оптически активного поликумарона открывает возможности для изготовления на его основе избирательных по оптической активности фильтров, адсорбирующих средств или ионообменных смол.
Получить оптически активные полимеры индена до сих пор не удалось. Таким образом, нет полной аналогии между строением полимеров индена и кумарона. Различия в структуре образующихся полимеров, а также в протекании процессов полимеризации кумарона и индена объясняются различными свойствами пятичленного цикла в том и другом случае.
2.3 Сополимеризация индена с кумароном и другими непредельными соединениями
В продуктах переработки каменноугольной смолы и сырого бензола инден и кумарон содержатся совместно. Поэтому особый интерес представляет исследование сополимеризации этих двух мономеров. Отметим, что в литературе нами найдены данные только по изучению катионного процесса.
В присутствии четырёххлористого титана реакция сополимеризации индена с кумароном протекает с хорошим выходом. При этом отмечено [23], что молекулярная масса закономерно понижается от 1,63 для полииндена до 0,28 дл/г с увеличением доли кумарона в смеси мономеров до 50 мол.%. Полимеризация в присутствии BF3 легко позволяет получить поликумарон и сополимер с 50 % индена с характеристической вязкостью 1,7 дл/г, что выше, чем с TiCl4.
Кроме индена и кумарона, в коксохимическом сырье могут содержаться также такие непредельные соединения, как стирол, a-метилстирол, циклопентадиен. В табл. 1.1 приводятся некоторые экспериментальные данные, характеризующие сополимеризацию этих соединений с инденом.
Таблица 1.1. - Условия сополимеризации индена со стиролом, a-метилстиролом, циклопентадиеном в хлористом метилене и вязкость (h) полученных сополимеров при выходе 100 % (по данным [23]).
| Второй мономер1) |
Катализатор2) |
Температура, ºС |
h, 100см3/г |
| Стирол |
TiCl4 |
-72 |
0,19 |
| BF3 |
-72 |
0,20 |
| BF3 |
-100 |
0,22 |
| a-метилстирол |
TiCl4 |
-72 |
0,54 |
| BF3 |
-97 |
1,45 |
| Циклопентадиен |
TiCl4 |
-72 |
0,44 |
| TiCl4 |
-93 |
0,85 |
1) концентрация 0,42 моль/л; 2) концентрация 0,02 моль/л.
При совместной полимеризации во всех случаях отмечается понижение молекулярной массы получаемого продукта. Особенно наглядно это видно на примере совместной полимеризации индена и стирола [23], если учесть, что в одинаковых условиях гомополимеризация стирола даёт продукт с вязкостью 0,56; a-метилстирола - 0,55; индена - 1,60 дл/г.
Сополимеризация кумарона со стиролом и a-метилстиролом при концентрации TiCl4 0,01 моль/л и температуре 201 К за 0,5 ч протекает с выходом 58 и 89 %, давая продукт, содержащий 48 и 44 % кумарона, с вязкостью 0,12 и 0,82 дл/г соответственно [23]. Таким образом, анализ имеющихся экспериментальных данных показывает, что катионная гомополимеризация и сополимеризация индена и кумарона наиболее активно протекает под действием таких катализаторов, как хлористый алюминий и трёхфтористый бор. Совместная полимеризация нескольких мономеров приводит обычно к получению полимеров с меньшей молекулярной массой, чем при их раздельной полимеризации. Снижению молекулярной массы способствует также применение ароматических растворителей.
2.4 Модификация инден-кумароновых смол
Требования многочисленных потребителей к качеству инден-кумароновых смол [16] весьма разнообразны и не все эти требования могут быть удовлетворены при существующей сырьевой базе и технологии производства смол. Недостаточная свето- и атмосферостойкость инден-кумароновых смол, хрупкость, неполная растворимость во многих полярных растворителях, несовместимость с синтетическими плёнкообразующими веществами и плохая совместимость с высыхающими маслами, невозможность получения термореактивных плёнкообразователей – эти свойства не удовлетворяют лакокрасочную промышленность. Не соответствует современным требованиям относительно высокое остаточное содержание в смоле легколетучих веществ, особенно в тех случаях, когда переработка инден-кумароновой смолы осуществляется при повышенной температуре.
Большие возможности для изменения качества смол даёт модификация их различными химическими соединениями. Помимо получения инден-кумароновых смол с требуемыми свойствами, модификация может довольно существенно увеличить ресурсы сырья для из производства.
Одним из способов придания инден-кумароновой смоле новых физико-механических свойств является приготовление разнообразных компаундов. Уже в этом простейшем случае речь идёт не о механической смеси нескольких соединений, а о возникновении нового вещества с отличающимися от образующих его компонентов свойствами.
Чаще встречается химическая модификация инден-кумароновой смолы, основывающаяся на том, что из-за наличия остаточной непредельности (по крайней мере одна двойная связь на молекулу полимера) смола обладает некоторой реакционной способностью и может присоединять различные соединения. Известно также, что в реакциях инден-кумароновой смолы могут участвовать активные водородные атомы метиленовых групп молекулы индена.
Модификация инден-кумароновых смол различными химическими веществами осуществляется как в расплаве, так и в растворах под воздействием инициаторов радикальной полимеризации или кислотных катализаторов ионной полимеризации.
Инденовая смола может непосредственно взаимодействовать с малеиновым ангидридом, образуя модифицированную карбоксилсодержащую углеводородную смолу, поликонденсацией которой с многоосновной кислотой (например, фталевой). Многоатомным спиртом (например, пентаэритритом) и высыхающим маслом далее получают алкидную смолу.
Модификация уже готовых инден-кумароновых смол вызывает необходимость дополнительной их обработки, что зачастую усложняет производство, а иногда приводит к ухудшению качественных показателей. С точки зрения удобства ведения процесса более целесообразно осуществлять совместную полимеризацию смолообразующих компонентов с модифицирующими добавками.
Хорошо известна способность индена и некоторых его гомологов образовывать аддукты с двухосновными кислотами, их ангидридами и эфирами. При этом как показали Хенглейн и Штольценбах на примере взаимодействия индена и малеинового ангидрида, продукт реакции имеет макромолекулярный характер (средняя молекулярная масса 30000 – 40000, при исследовании ММР показано наличие фракции с молекулярной массой 100000). Реакция протекает по двойным связям индена и малеинового ангидрида с получением продукта линейной структуры [16]:
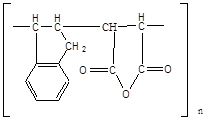
Для придания полученному сополимеру плёнкообразующих свойств [16] проводили дальнейшую его модификацию путём алкоголиза ангидридных связей с получением кислых эфиров следующего строения:
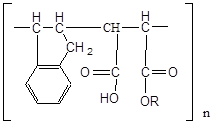
Для улучшения эластичности плёнок составляли композиции полученных продуктов с гликолями.
Наряду с индивидуальными химическими соединениями для получения модифицированных инден-кумароновых смол могут быть привлечены различные технические продукты, богатые непредельными соединениями. Количество модифицирующего агента в этом случае можно варьировать в широком диапазоне, что позволяет существенно повысить выход смолы.
2.5 Основные направления использования инден-кумароновых смол
Инден-кумароновые смолы [16] обладают очень ценными свойствами: водостойкостью и устойчивостью к кислотам и щелочам, высокой связующей и клеящей способностью, способностью образовывать прочную плёнку, малой электро- и теплопроводностью, способностью образовывать эмульсии, в ряде случаев – также способностью совмещаться с высыхающими и полувысыхающими маслами. Будучи к тому же сравнительно недорогими, инден-кумароновые смолы находят широкое применение в различных отраслях промышленности.
2.5.1 Производство лаков, красок и защитных покрытий
В лаках и красках инден-кумароновые смолы играют роль плёнкообразующих веществ [16]. Для приготовления масляно-смоляных лаков инден-кумароновую смолу обычно смешивают с высыхающими растительными маслами, после чего полученный продукт совмещения для удобства нанесения покрытия растворяют в подходящем растворителе, чаще всего в уайт-спирите. В процессе совмещения инден-кумароновой смолы с растительными маслами происходит, по-видимому, сополимеризация.
Лаки на основе инден-кумароновых смол успешно используют для получения коррозионно-стойких покрытий на коксохимических заводах. Специальная область применения инден-кумароновых смол – электроизоляционные лаки и диэлектрические замазки. Добавляя 20 % инден-кумароновых смол к каучукам, в США получают смеси с хорошими изоляционными свойствами, которые используют для защиты кабелей.
Кроме приготовления лаков с высыхающими маслами инден-кумароновые смолы используют для получения твёрдых лаков в комбинации с алкидными смолами и силиконовыми резинами. На их основе готовится, например, термостойкий серебряный лак, содержащий в своём составе инденовую смолу, силиконовую резину и ароматические растворители.
Иногда инден-кумароновую смолу очень успешно применяют [16] при изготовлении смешанных олиф самого различного состава, добавляя её взамен канифоли. Лаки, приготовленные на кумароновой смоле, не уступают канифольным лакам и могут использоваться для изготовления эмалевых красок (тёмных цветов), а также для разведения густотёртых красок без ухудшения их цвета.
2.5.2 Производство резино-технических изделий
В производстве резины инден-кумароновая смола играет роль размягчителя и реже роль разбавителя (экстендера) для частичной замены каучука [16]. Мягчители способствуют более равномерному распределеиню ингредиентов в резиновой смеси, в частности улучшают диспергирование в смеси наполнителей, что приводит к повышению эластичности резиновой смеси и улучшению физико-механических свойств вулканизатов. Добавка инден-кумароновой смолы, кроме того, повышает клейкость резиновых смесей.
Инден-кумароновые смолы применяют [16] в качестве мягчителей как синтетического, так и натурального каучука при производстве резин различного назначения, включая шинные резины. Жидкие смолы служат для пропитки корда в производстве шин.
2.5.3 Производство отделочных материалов, клеев, мастики.
Одним из наиболее крупных потребителей инден-кумароновых смол в настоящее время является промышленность строительных материалов, где смолы используются в качестве связующего при производстве плиток для полов, линолеума и других отделочных материалов [16]. По виду связующей плитки делятся на кумароновые и кумаронохлорвиниловые. Плитки применяют главным образом для настилки полов в жилых, общественных и промышленных зданиях с сухими процессами производства при отсутствии интенсивного механического воздействия на пол. Кроме связующего, в состав плиток вводят пластификатор, наполнитель и пигмент. Помимо плиток, инден-кумароновая смола может входить в состав рулонных облицовочных материалов типа линолеума.
Применение инден-кумароновых смол в производстве вспененного поливинилхлорида [16] при повышенной температуре создаёт условия для лучшего вспенивания, что способствует образованию объёмной ячеистой структуры. Инден-кумароновую смолу применяют также при изготовлении мастики, используемой для приклеивания облицовочных материалов. Так, кумарон-каучуковая мастика, изготовленная из инден-кумароновой смолы, модифицированной наиритом, характеризуется высокой адгезией к поливинилхлориду, большой прочностью приклейки и быстрым нарастанием прочности. Мастики обычно представляют собой пасту, составленную из битума, инден-кумароновой смолы, наполнителей и растворителей. Применяют мастики также для изготовления наливных полов.
Инден-кумароновую смолу широко используют для изготовления обмазочных и шпаклёвочных материалов [16], а также клеев, которым она придаёт водостойкость [16]. Для увеличения прочности склеивания инден-кумароновая смола вводится в состав контактных клеев, заменяющих механический способ крепления (болты, винты, гвозди).
2.5.4 Получение антикоррозионных материалов
Неблагоприятная экологическая ситуация в регионе Донбасса [29, 30] и в целом в Украине вызвана возрастающими масштабами накопления промышленных отходов. Загрязнение окружающей среды при повышении его агрессивности наносит огромный ущерб экономике страны. В частности, в результате коррозии сталебетонных и металлических конструкций нередки аварийные ситуации, которые усугубляют опасность возникновения экологической обстановки с несчастными исходами. В связи с этим поиск способов утилизации отходов в качестве противокоррозионных материалов, в том числе ингибиторов коррозии и защитных покрытий, одновременно снизит экологическую напряжённость и даст экономический эффект в результате ресурсосбережения и расширения сырьевой базы народнохозяйственного комплекса.
Инден-кумароновые смолы, получаемые из коксохимического сырья, обладают водостойкостью и устойчивостью к кислотам и щелочам, способностью образовывать прочную пленку, благодаря чему лаки на их основе успешно используют для получения коррозионно-стойких покрытий на коксохимических заводах [30, 31]. С использованием инден-кумароновых смол, полученных полимеризацией смолообразующих компонентов тяжёлой фракции бензола при переработке каменного угля, а также кубовых остатков (фракция С20 и выше) синтетических жирных кислот, полученных при переработке нефти, также разработаны противокоррозионные материалы [
29
]. В качестве ингибирующих добавок к ним использованы кубовые смолистые остатки производства оксиамина Крымского ПО «Химпром», являющиеся смесью первичных, вторичных и третичных нитроаминов. Введение в противокоррозионные композиции на основе инден-кумароновой фракции амино-содержащих смолистых отходов повышают защитные свойства плёнок [30].
Малая электропроводность инден-кумароновых смол позволяет применять их в составе электроизоляционных лаков и диэлектроческих замазок, в том числе в смеси с каучуками [32]. В производстве резины инден-кумароновая смола играет роль мягчителя, улучшает диспергирование наполнителей в резиновой смеси, что приводит к повышению ее эластичности и улучшению физико-механических свойств вулканизатов [31].
Одним из наиболее крупных потребителей инден-кумароновых смол может быть промышленность строительных материалов, где возможно их использование в качестве связующего при производстве плиток для полов, линолеума и других отделочных материалов [14,32,33]. Кроме того, эти смолы могут применяться как связующее при получении формовочных композиций из древесных отходов [34], в производстве углеграфитовых изделий [35=4], для изготовления дорожных пластобетонов [36], в качестве адгезивов для асфальта [37].
Как видно из этого краткого перечня, разнообразие свойств инден-кумароновых смол, их сравнительная дешевизна способствуют тому, что эти полимеры могут быть востребованы различными потребителями. Однако при получении их наиболее распространенным методом катионной полимеризации трудности, связанные с тщательным отделением катализатора, отрицательно влияют на их качество, могут вызвать подпленочную коррозию, снижают водостойкость покрытия. Термическая полимеризация не имеет этих недостатков, однако процесс термополимеризации чрезвычайно длителен (более 72 ч), дают невысокий выход (~ 60 %), требует больших энергетических затрат (температура процесса 230-250°С). Одним из способов, позволяющим регулировать качество инден-кумароновых смол и уменьшить отрицательные стороны катионной и термополимеризации, является модификация смол. Одним из возможных модификаторов является малеиновый ангидрид, который способен к активному взаимодействию с непредельными соединениями, входящими в состав кумарон-инденовой фракции.
2.5.5 Другие области применения инден-кумароновых смол
Кроме названных выше областей потребления, инден-кумароновую смолу используют [16] при производстве синтетической кожи и кожзаменителей, гидроизоляционных материалов. Пластобетона, для пропитки бумаги и тканей, при получении шлифовальных паст, при производстве пестицидов, ионообменных смол, для приготовления охлаждающих эмульсий для металлорежущих станков, в качестве эмульгатора для эмульсионной полимеризации. Инден-кумароновая смола может найти применение [16] при изготовлении противопригарной упрочняющей краски для литейных форм и стержней, в качестве присадки, понижающей температуру застывания минеральных масел, для улучшения закалочных свойств (квенчинг-эффекта) нафтеновых или парафиновых масел, в составе рецептуры фунгицидной замазки для обработки деревьев, для антисептической пропитки рыболовных сетей, для изготовления эмульсий, предупреждающих эрозию почвы, для придания жёсткости клеевому составу на основе каучука, используемому для связывания щебня или гравия при установке балластного слоя под железнодорожную колею.
Инден-кумароновые смола может использоваться [16] в качестве связующего при получении формовочных композиций из древестных отходов, а также для склеивания слюды и приклеивания её к подложке. На основе инден-кумароновых смол готовят звукопоглощающие пластмассовые композиции, вяжущее для изготовления дорожных пластобетонов, адгезивы для асфальта. Возможно также использовать инден-кумароновую смолу в качестве связующего и пропитывающего материала в производстве углеграфитовых изделий, растёт использование инден-кумароновых смол в отделочных и других строительных материалах.
3. Методика эксперимента
3.1 Исходные вещества и их физико-химические характеристики
Таблица 3.1 – Характеристика веществ, используемых в работе
| Название |
Mr |
Плотность, г/см3 |
Показатель преломления |
Температура, К |
Формула |
| d20 |
d60 |
Tкип. |
Tпл |
| Инден |
116,15 |
0,996 |
- |
1,5768 |
455,6 |
275,6 |
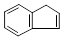 |
| Малеиновый ангидрид |
98,06 |
1,48 (_б.) |
1,314 |
1,6092 |
- |
333,15 |
 |
| Толуол |
92,14 |
0,867 |
- |
1,4969 |
383,8 |
178,15 |
С6Н5СН3 |
| Ацетон |
58,08 |
0,791 |
- |
1,3591 |
329,39 |
177,8 |
(СН3)2СО |
| Хлороформ |
119,38 |
1.488 |
- |
1,4455 |
334,30 |
209.65 |
CHCl3 |
| ТГФ |
72,11 |
0,889 |
- |
1,4050 |
338,75- 339,75 |
208,15 |
(CH2)4O |
| 1,4-Диоксан |
88,10 |
1,3375 |
- |
1,4224 |
374,47 |
284,95 |
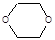 |
| ДМСО |
78,13 |
1,1014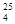 |
- |
1,477025 |
462,15 |
291,6 |
СН3SOСН3 |
| Петролейный эфир |
- |
0,645 – 0,66515 |
- |
1.365 |
313-338 |
- |
- |
3.2 Методика очистки мономеров и растворителей
Малеиновый ангидрид очищали возгонкой. Инденовую фракцию выделяли при перегонке кумарон-инденовой фракции Макеевского КХЗ.
Толуол очищали методом физической осушки от воды с последующей перегонкой при атмосферном давлении. Хлороформ сушили над СаCl2, а затем перегоняли. Ацетон сушили над K2CO3 и перегоняли.
Тетрагидрофуран очищали, удаляя следы пероксидов кипячением 0,5%-ной суспензии Cu2Cl2 в тетрагидрофуране в течение 30 мин с последующей перегонкой при атмосферном давлении. Затем тетрагидрофуран сушили над гранулами КОН, кипятили с обратным холодильником и снова перегоняли.
1,4-диоксан выдерживали несколько суток с гранулами КОН после чего перегоняли над свежей щёлочью. ДМСО, выдержанный над ВаО, перегоняли в вакууме над гранулами NaOH.
3.3 Методика синтеза полимера
В предварительно взвешенную на аналитических весах колбу с помощью градуированной пипетки вносим инденовую фракцию, содержащую ~ 85 % чистого индена (~ 90 мол. %). Рассчитываем массу малеинового ангидрида (МА), соответствующую 10 мол. % от массы взятого индена; тетрабутоксититана, соответствующую 5 мол. % МА, и пероксида бензоила (ПБ). Добавляем и взвешиваем растворитель – диоксан или толуол. Количество ДО рассчитываем, чтобы суммарная концентрация мономеров в растворе составляла 3 моль/л, а толуол берём в объёме, равном суммарному объёму мономерной смеси. После взвешивания рассчитанного количества пероксида бензоила вносим его в колбу с инденовой фракцией и растворителем. Затем берём рассчитанное количество МА и Ti(Obu)4 и вносим эти компоненты в колбу с реакционной смесью. Реакционную смесь заливаем в дилатометр или ампулы. Если процесс проводится в ампулах, их взвешиваем до и после наполнения реакционной смесью, затем запаиваем и взвешиваем, после чего помещаем ампулы в термостат. Отсчёт времени полимеризации начинаем через 10 мин с момента погружения ампул. При проведении полимеризации в дилатометре время отмечаем с момента достижения максимального объёма смеси, после чего следим за изменением объёма с помощью катетометра В-630.
По истечении времени полимеризации вынимаем дилатометр или ампулы из термостата и охлаждаем их. Выливаем жидкую фазу из дилатометра в предварительно взвешенный бюкс и снова взвешиваем, после чего используем для высаждения. Раствор из бюкса или ампул постепенно переносим в химический стакан с петролейным эфиром (50 см3) при постоянном перемешивании. Если в ампуле есть твёрдый остаток, его растворяем в том же растворителе (~5 см3) и также выливаем в петролейный эфир. Отфильтровываем полученный осадок через взвешенный фильтр Шотта при помощи водоструйного насоса, предварительно смочив фильтр растворителем. После фильтрования полимер сушим в вакуум-эксикаторе, периодически взвешивая его до постоянной массы. Выход полимера ω (%) рассчитываем по формуле:
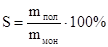 (3.1) (3.1)
где S – выход полимера; 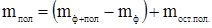 – масса полимера, – масса полимера, 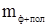 - масса фильтра с полимером, - масса фильтра с полимером, 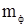 - исходная масса фильтра, - исходная масса фильтра, 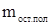 - масса остатка полимера в стакане; - масса остатка полимера в стакане; 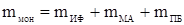 – масса мономеров, – масса мономеров, 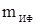 - масса инденовой фракции, - масса инденовой фракции, 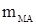 - масса малеинового ангидрида, - масса малеинового ангидрида, 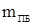 -масса пероксида бензоила. -масса пероксида бензоила.
3.4 Методика определения характеристической вязкости
В предварительно взвешенную на аналитических весах колбу 1 с пришлифованной пробкой вносим сополимер индена с малеиновым ангидридом. Затем добавляем 8 см3 растворителя и снова взвешиваем. Выдерживаем раствор в закрытой колбе до растворения полимера и заливаем в вискозиметр отмеренную с помощью пипетки аликвоту через фильтр Шотта. Если полимер не растворился, фильтруем раствор через предварительно взвешенный фильтр Шотта в предварительно взвешенную колбу 2. Колбу 1 после полного испарения растворителя взвешиваем для определения потери полимера в колбе.
Далее таким же образом определяем количество полимера на фильтре Шотта. Взвесив колбу 2 с отфильтрованным раствором, уточняем его количество после фильтрования.
Начальную концентрацию раствора Со рассчитываем по формуле:
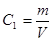 , (3.2) , (3.2)
где m – масса навески полимера в колбе,
V – объём раствора.
С помощью вискозиметра Уббелодде, термостатированного при температуре 30+0,1º С определяем время истечения чистого растворителя или раствора полимера, проведя по 5 измерений.
После измерения времени истечения исходного раствора полимера последовательно добавляем к раствору в вискозиметре порции растворителя и измеряем время истечения полученного раствора.
Текущие концентрации полимера в растворе рассчитываем по формуле:
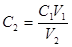 , (3.3) , (3.3)
где V1 – начальный объём раствора,
V2 – объём раствора, соответствующий концентрации С2.
По времени истечения растворов рассчитываем их относительную, удельную и приведённую вязкость Относительная вязкость равна
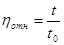 , (3.4) , (3.4)
где t – время истечения раствора, с, t0 – время истечения чистого растворителя, с.
Удельную и приведённую вязкость рассчитываем по формулам:
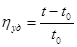 , (3.5) , (3.5)
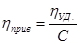 , (3.6) , (3.6)
Затем строим графическую зависимость ηприв – С и методом экстраполяции на ось ординат определяем характеристическую вязкость.
3.5 Методика статистической обработки результатов определения времени истечения
Для оценки ошибки измерения рассчитываем следующие статистические характеристики результатов эксперимента:
выборочное среднее:
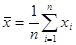 , (3.7) , (3.7)
где xi – результаты параллельных измерений, n – число параллельных измерений (наблюдений, анализов).
Исправленная выборочная дисперсия 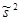 : :
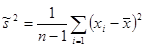 . (3.8) . (3.8)
выборочное среднеквадратическое или стандартное отклонение
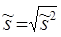 (3.9) (3.9)
доверительный интервал для среднего значения измеряемой величины 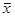
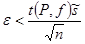 , (3.10) , (3.10)
где 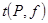 - значение t-распределения Стъюдента при числе степеней свободы - значение t-распределения Стъюдента при числе степеней свободы 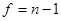 и уровне значимости и уровне значимости 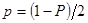 . .
4. Техника безопасности
4.1 Требования безопасности при работе со стеклянной посудой и приборами
При получении новой посуды и перед каждым использованием необходимо её тщательно осмотреть. Изделия, имеющие изъяны, нельзя использовать для работы.
Осколки разбитой посуды убираются с помощью щётки и совка, а не руками.
4.2 Работа со стеклянными ампулами
В стеклянные ампулы разрешается запаивать сконденсированные газообразные вещества, имеющие температуру кипения не ниже 120 С, заполнять ампулы не более чем на 50 % их объёма. Запрещается запаивать в ампулу вещества, при нагревании разлагающиеся со взрывом. Запаянные ампулы вскрывают только после охлаждения их ниже температуры кипения запаянного в них вещества. После охлаждения ампулы заворачивают в полотенце, затем слегка делают надрез напильником на капилляре и отламывают его. При вскрытии запаянный конец направляется в сторону, где нет людей.
4.3 Требования безопасности при проведении нагревания
Источниками опасности являются газовые горелки, электронагревательные приборы, высокая температура в рабочей зоне, присутствие в зоне нагрева ЛВЖ и ГЖ.
Запрещается использование в лаборатории электрических плиток с открытой спиралью. Для нагрева ЛВЖ и ГЖ необходимо использовать жидкостные бани с диаметром не менее диаметра нагревательного элемента плитки. В качестве теплоносителей допускается использовать только чистые жидкости, не содержащие посторонних примесей и загрязнений.
Запрещается нагревание жидкостей в закрытых колбах или приборах, не имеющих сообщения с атмосферой. Подобные работы проводятся либо в лабораторных автоклавах, либо в стеклянных толстостенных ампулах. Эти работы относятся к категории особоопасных и требуют разработки специальных инструкций по охране труда.
4.4 Мытьё посуды
Мыть посуду необходимо в течение рабочего дня, не накапливая грязную посуду на рабочем столе. В вытяжном шкафу или в общей лабораторной раковине. Грязную посуду следует складывать в специальные кюветы.
При мытье посуды надо надевать резиновые перчатки, а в случае использования агрессивных жидкостей (хромовая смесь, концентрированные щёлочи и т.п.) – защитные очки или маску, прорезиненный или полиэтиленовый фартук.
Первичное ополаскивание посуды, загрязнённой легколетучими, вредными или дурнопахнущими веществами, следует производить в вытяжном шкафу.
Мытьё органическими растворителями (этиловый спирт, ацетон, хлороформ и др.) проводят в вытяжном шкафу вдали от нагревательных приборов. Ополаскивая посуду изнутри несколько раз минимальными порциями подходящего растворителя, сливая их в специально отведенную для этого банку (слив).
С органическими веществами и растворителями хромовая смесь может иногда реагировать со взрывом. Поэтому перед мытьём хромовой смесью посуду очищают с помощью горячей воды и ерша. Необходимо тщательно удалить смазку со шлифов. Малую посуду погружают в хромовую смесь целиком на 20 минут, крупную ополаскивают небольшим количеством хромовой смеси, сливая её обратно в сосуд для хранения. Через 20 минут моют посуду тёплой водой. Для выемки посуды из хромовой смеси используют тигельные щипцы.
Для приготовления безводной хромовой смеси к 100 мл концентрированной серной кислоты добавляют 10 г тонкоизмельчённого бихромата калия или натрия (смешивание производят в фарфоровом стакане). При приготовлении и работе с хромовой смесью следует помнить, что безводная хромовая смесь опаснее концентрированной серной кислоты. Образующийся в хромовой смеси оксид хрома (ПДК=0,01 мг/м3) является едким летучим соединением.
Запрещается использовать для приготовления хромовой смеси азотную кислоту ввиду чрезвычайно высокой опасности такой смеси.
Запрещается применение смеси концентрированных азотной и серной кислот для растворения смолистых органических загрязнений на химической посуде: возможен взрыв из-за образования сульфинатов или полинитросоединений.
После окончательного ополаскивания для стекания воды посуду помещают на колках сушилки, затем помещают в сушильный шкаф, не допуская прикосновения холодной посуды к горячей. Вынимают посуду остывшей после выключения шкафа.
4.5 Требования безопасности при работе с легковоспламеняющимися жидкостями
К легковоспламеняющимся жидкостям относятся вещества с температурой вспышки (наименьшая температура горючего вещества, при которой над его поверхностью образуются пары и газы, способные вспыхнуть в воздухе от внешнего источника зажигания) ниже 610 С.
Все легковоспламеняющиеся жидкости (ЛВЖ) являются токсичными веществами; многие из них при комнатной температуре образуют пероксиды, с воздухом – пожаро- и взрывоопасные смеси; они легко воспламеняются, быстро горят и с трудом тушаться.
Разряды опасности ЛВЖ:
I разряд опасности – особоопасные ЛВЖ (с температурой вспышки до – 180 С);
II разряд опасности – постоянно опасные ЛВЖ (с температурой вспышки от – 18 до 23 0 С);
III разряд опасности –ЛВЖ, опасные при повышенной температуре (с температурой вспышки от 23 до 610 С);
Общий запас одновременно хранящихся в каждом рабочем помещении ЛВЖ и ГЖ рассчитывается с учётом того, чтобы образующаяся в случае аварии взрывоопасная смесь горючих газов и паров не превышала 5 % объёма воздуха в помещении. Обычно рекомендуется хранить в одностандщартной комнате (объём помещения – 100 м3) не более 5л ЛВЖ, в двухстандартной не более 10 л и т.д.
Общее количество огнеопасных жидкостей в рабочем помещении не должно превышать суточной потребности, но не более 2 – 3 л на 1 сотрудника. Суточные нормы ЛВЖ и ГЖ утверждаются руководителем структурного подразделения по согласованию с инженером по охране труда. ЛВЖ и ГЖ (за исключением веществ, имеющих низкую температуру кипения) должны храниться в лабораторном помещении в толстостенных банках (склянках) с притёртыми пробками. Запрещается хранить ГЖ в полиэтиленовой, а также тонкостенной стеклянной посуде ёмкостью более 200 мл.
4.5 Техника безопасной работы с пероксидом бензоила
Перекристаллизацию пероксида бензоила ни в коем случае нельзя проводить при нагревании. Перекристаллизация его из горячего хлороформа опасна. Температуру его плавления определяют только в крайнем случае. Для очистки можно брать не более двух грамм пероксида бензоила.
5. Экспериментальная часть
5.1 Изучение сополимеризации инденовой фракции с малеиновым ангидридом
Изучение скорости накопления сополимера проводили в растворе ДО без и в присутствии комплексообразователя (смеси I и II). Состав исходной мономерной смеси с суммарной концентрацией мономеров 3 моль/л и пероксида бензоила 0,03 моль/л приведен в таблице 5.1.
Таблица 5.1 – Состав реакционной смеси
| Компонент |
Масса, г |
Объём компонентов
при 60о С, мл
|
| Смесь I |
Смесь II |
Смесь I |
Смесь II |
| 1 |
2 |
3 |
4 |
5 |
ИФ,
в т. ч. инден
|
3,68947
3,13605
|
3,74780
3,18563
|
3,860
3,147
|
3,921
3,196
|
| ПБ |
0,07300 |
0,08145 |
0,055 |
0,061 |
| МА |
0,29430 |
0,29845 |
0,224 |
0,227 |
| 1,4-диоксан |
6,02425 |
6,05335 |
6,097 |
6,127 |
| Тетрабутоксититан |
- |
0,02160 |
- |
- |
| Всего |
10,08102 |
10,20265 |
10,236 |
10,336 |
| В т.ч. инден+МА |
3,43035 |
3,48408 |
3,371 |
3,423 |
Реакционную смесь вылили в откалиброванный дилатометр и следили за изменением объёма в процессе сополимеризации. Объём смеси в дилатометре, согласно откалиброванной величине Vдил и начальному значению метки Vo составил 9,1765 и 9,0815 мл.
Исходя из объёма мономеров в реакционной смеси находим для опытов 1 и 2 (смеси I и II):
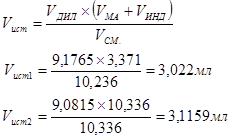
Результаты изменения объёма для изучения процесса сополимеризации смесей I и II приведены в табл. 5.2.
Таблица 5.2 – Данные кинетических измерений для реакции сополимеризации инденовой фракции с малеиновым ангидридом в соотношении 90:10 мольн. % в ДО (60 °С)
| Смесь I |
Смесь II |
| t, мин |
V,мл |
ΔV,мл |
ΔV/Vист., •100 % |
S, % |
t, мин |
V, мл |
ΔV,мл |
ΔV/Vист., •100 % |
S, % |
| 0,00 |
0,490 |
0,000 |
0,000 |
0,00 |
0,00 |
0,585 |
0,000 |
0,000 |
0,00 |
| 21,00 |
0,495 |
0,005 |
0,158 |
1,14 |
11,75 |
0,590 |
0,005 |
0.160 |
1,08 |
| 35,80 |
0,505 |
0,015 |
0.473 |
3,14 |
16,20 |
0,595 |
0,010 |
0.321 |
2,17 |
| 40,80 |
0.510 |
0,020 |
0,631 |
4,54 |
31,55 |
0,600 |
0,015 |
0,481 |
3,25 |
| 55,20 |
0,515 |
0,025 |
0,789 |
5,68 |
46,45 |
0,605 |
0,020 |
0,642 |
4,34 |
| 67,80 |
0,520 |
0,030 |
0,946 |
5,90 |
58,75 |
0,610 |
0,025 |
0,802 |
5,42 |
| 79,40 |
0.523 |
0,033 |
1,041 |
7,49 |
73,90 |
0,615 |
0,030 |
0,963 |
6,51 |
| 82,80 |
0,525 |
0,035 |
1,104 |
7,94 |
94,12 |
0,620 |
0,035 |
1,123 |
7,59 |
| 85,70 |
0.527 |
0,037 |
1,167 |
8,40 |
124,87 |
0,630 |
0,045 |
1,444 |
9,76 |
| 95,85 |
0.530 |
0,040 |
1,262 |
9,08 |
134,90 |
0,635 |
0,050 |
1,605 |
10,84 |
| 106,50 |
0,533 |
0,043 |
1,357 |
9,76 |
138,01 |
0,640 |
0,055 |
1,765 |
11,93 |
| 109,90 |
0,535 |
0,045 |
1,420 |
10,22 |
156,20 |
0,645 |
0,060 |
1,926 |
13,01 |
| 125,50 |
0,540 |
0,050 |
1,578 |
11,35 |
188,71 |
0,650 |
0,065 |
2,086 |
14,09 |
| 143,10 |
0,545 |
0,055 |
1,735 |
12,48 |
213,50 |
0,655 |
0,070 |
2,247 |
15,18 |
| 168,50 |
0,550 |
0,060 |
1,893 |
13,62 |
240,00 |
0,660 |
0,075 |
2,407 |
16,26 |
| 182,50 |
0,555 |
0,065 |
2,051 |
14,76 |
| 206,00 |
0,560 |
0,070 |
2,209 |
15,89 |
| 227,30 |
0,565 |
0,075 |
2,367 |
17,03 |
| 266,75 |
0,570 |
0,080 |
2,524 |
18,16 |
Выход высажденного сополимера для смесей I и II составил 18,16 и 16,26 мас. % соответственно. На основании этих величин и соответствующих им значений 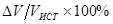 , равных 2,524 и 2,407, находим величину коэффициента контракции k, как соотношение и S: , равных 2,524 и 2,407, находим величину коэффициента контракции k, как соотношение и S:
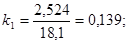 . .
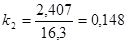
На основании полученных значений были построены зависимости S от t, приведенные на рис. 5.1 и 5.2.

Рисунок 5.1 – Зависимость накопления полимера (S) от времени для полимеризации смеси I в 1,4-диоксане при 60оС.
Для изучения влияния растворителя и гетерогенности при том же соотношении ИФ и МА была проведена полимеризация в растворе толуола и диоксана. Реакционную смесь залили в шесть ампул, три из которых были наполнены раствором исходных компонентов в диоксане, а три – раствором в толуоле. Состав реакционной смеси приведен в таблице 5.3, а распределение её по ампулам и выход полимеров – в табл. 5.4.
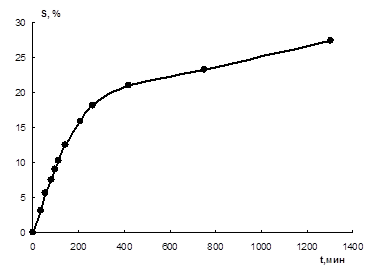
Рисунок 5.2 – Зависимость накопления полимера от времени для полимеризации смеси II при 60оС (ДО).
Таблица 5.3 – Состав реакционной смеси для полимеризации ИФ и МА до высоких конверсий при 600 С
| Компонент |
Масса компонентов (г) в смеси |
Доля компонета (масс. %) |
| Смесь I’ |
Смесь III |
Смесь I’ |
Смесь III |
ИФ, в
т. ч. инден
|
1,85209
1,57428
|
1,45934
1,24044
|
2,15042
1,82786
|
1,72768
1,46853
|
| МА |
0,14851 |
1,24044 |
0,16936 |
0,13606 |
| ПБ |
0,04036 |
0,11702 |
0,04968 |
0,04006 |
| 1,4 – диоксан |
3,33828 |
0,03180 |
- |
- |
| Толуол |
- |
2,71517 |
2,15536 |
1,73165 |
| Всего |
5,37924 |
4,32333 |
4,52482 |
3,63545 |
| В т. ч. мономеров |
1,72279 |
1,35746 |
1,99722 |
1,60459 |
Таблица 5.4 – Содержание реакционной смеси в ампулах и выход сополимеров при полимеризации смесей I’ и III
| № ампулы |
Масса, г |
Время полимеризации, мин |
Масса выделенного полимера, г |
Выход, % |
| реакционной смеси |
мономеров |
| 1* |
5,38085 |
1,72279 |
420 |
0,32995 |
19,15 |
| 2* |
5,63210 |
1,80323 |
750 |
0,37890 |
21,01 |
| 3* |
4,23980 |
1,35746 |
1305 |
0,33665 |
24,80 |
| 4** |
4,49220 |
1,99722 |
420 |
0,16385 |
8,34 |
| 5** |
3,60910 |
1,60459 |
750 |
0,19895 |
12,40 |
| 6** |
2,79810 |
1,24402 |
1305 |
0,32595 |
26,20 |

Рисунок 5.3 – Зависимость «S – t» для полимеризации ИФ с МА в ДО
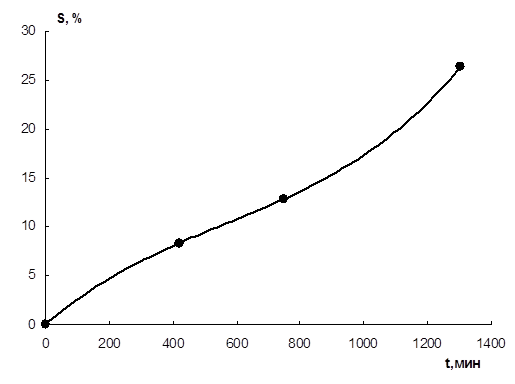
Рисунок 5.4 – Зависимость «s – t» для полимеризации ИФ с МА в толуоле (смесь III) при 60º С
Зависимость «S – t» для полимеризации ИФ с МА в ДО и толуоле приведена на рис. 5.3. и 5.4.
Чтобы выяснить влияние температуры полимеризации на скорость процесса и свойства сополимера, провели процесс при повышении температуры от 80о до 100о С. При этом смесь IV прогревали ~0,5 ч, поднимая температуру от 80 до 90оС, выдерживали при 90оС 3 ч, затем нагревали до 100о и выдерживали ещё 2 ч. В таком же режиме и с теми же катализаторами проводили реакцию исходной КИФ с МА (смесь V). Состав смеси и их содержание в ампулах, а также выход полимеров приведен в табл. 5.5 и 5.6.
Таблица 5.5 – Состав реакционной смеси для полимеризации ИФ и МА в ампулах при температуре 80 – 1000 С
| Смесь IV |
Смесь V |
| Компонент |
Масса, г |
Масс. доля, % |
Компонент |
Масса, г |
Масс. доля, % |
ИФ
В т. ч. инден
|
4,68540
3,98260
|
47,82
40,65
|
КИФ
в т. ч. активная часть
|
10,8615
4,3446
|
95,09
38,03
|
| МА |
0,37950 |
3,87 |
МА |
0,41655 |
3,65 |
| ПБ |
0,05155 |
0,53 |
ПБ |
0,1084 |
0,95 |
| ДТБП |
0,06175 |
0,63 |
ДТБП |
0,12485 |
0,09 |
| Ti(Obu)4 |
0,02665 |
0,27 |
Ti(Obu)4 |
0,03635 |
0,31 |
| Толуол |
4,59350 |
46,88 |
Всего |
11,4228 |
100,00 |
| Всего |
9,79835 |
100,00 |
5.2 Определение характеристической вязкости полученных продуктов
Вязкость растворов сополимеров индена с малеиновым ангидридом была измерена в хлороформе, ацетоне и ТГФ. Результаты определения приведены в таблицах 5.6, 5.7, 5.8 и 5.9.
По данным табл. 5.6 построены зависимости приведённой вязкости от концентрации растворов сополимера в хлороформе и ацетоне, приведенные на рис. 5.5 и 5.6. Сополимеры, полученные в ДО (смеси I и II, табл. 5.1), как было выяснено, с трудом растворимы в ТГФ и нерастворимы в ацетоне. Поэтому для них определение вязкости было проведено только в растворе ДМСО. Результаты этих определений приведены в табл. 5.6, 5.7 и 5.8 и на рис. 5.9.
Таблица 5.6 – Данные определения вязкости растворов сополимера индена с малеиновым ангидридом в хлороформе и ацетоне. Условия сополимеризации: [ПБ]=2 мас. %, 80 ºС, в растворе толуола (СПЛ 1)
| Растворитель |
Объём раствора, мл |
Концентрация раствора, г/дл |
Среднее время истечения, с |
ηотн |
ηуд |
ηуд/С., дл/г |
| Хлороформ |
7 |
0,0555 |
84,5 |
1,109 |
0,109 |
1,982 |
| 9 |
0,043 |
83,2 |
1,091 |
0,091 |
2,068 |
| 12 |
0,0323 |
82,6 |
1,083 |
0,083 |
2,441 |
| 16 |
0,0243 |
81,7 |
1,071 |
0,071 |
2,731 |
| Ацетон |
8 |
0,8503 |
93,76 |
1,080 |
0.080 |
0,094 |
| 10 |
0,6803 |
92,71 |
1,068 |
0,068 |
0,095 |
| 13 |
0,5233 |
91,75 |
1,057 |
0,057 |
0,109 |
| 6 |
0,9001 |
90,82 |
1,046 |
0,046 |
1,1108 |
| 7 |
0,0870 |
87,54 |
1,0082 |
0,0082 |
0,0943 |
| 9 |
0,0677 |
87,35 |
1,0060 |
0,0060 |
0,0886 |
| 12 |
0,0508 |
87,34 |
1,0059 |
0,0059 |
0,1161 |

Рисунок 5.5 – Зависимость приведённой вязкости от концентрации раствора сополимера в хлороформе для СПЛ 1. Условия сополимеризации: [ПБ]=2 мас. %, 80 ºС, в растворе толуола

Рисунок 5.6 – Зависимость приведённой вязкости от концентрации растворов сополимера в ацетоне в соотношении инден: МА 90:10 (без комплексообразователя)
Таблица 5.7 – Данные определения вязкости растворов сополимера индена с малеиновым ангидридом в ацетоне и ТГФ. Условия сополимеризации: [ПБ] = 2 мас. %, в толуоле 80 ºС (СПЛ 2)
| Растворитель |
Объём жидкости, мл |
Концентрация раствора, г/дл |
Время истечения, с |
ηотн |
ηуд |
ηуд/С., дл/г |
| Ацетон |
8 |
0,7798 |
96,09 |
1,107 |
0,107 |
0,137 |
| 10 |
0,6238 |
95,28 |
1,097 |
0,097 |
0,155 |
| 13 |
0,4798 |
93,32 |
1,075 |
0,075 |
0,156 |
| 17 |
0,3669 |
91,85 |
1,058 |
0,058 |
0,158 |
| Тетрагидрофуран |
8 |
0,8006 |
132.57 |
1,152 |
0,152 |
0,190 |
| 10 |
0,6405 |
127,87 |
1,111 |
0,111 |
0,173 |
| 13 |
0,4927 |
126,29 |
1,097 |
0,097 |
0,197 |
| 17 |
0,3768 |
123,21 |
1,070 |
0,070 |
0,186 |
На основании данных таблицы 5.7 построена зависимость ηприв – С, приведенная на рис. 5.7.
Данные определения вязкости для СПЛ 3 приведены в табл. 5.8.

Рисунок 5.7 – Зависимость ηприв – С, растворов в ацетоне (1) и тетрагидрофуране (2) (СПЛ 2)
Таблица 5.8 – Данные определения вязкости растворов сополимера индена с малеиновым ангидридом в ацетоне. Соотношение инден : МА 57:43. Условия получения: [ПБ] = 2 мас. %, [Ti(BuO)4]=5 мольн %, в толуоле, 80ºС (СПЛ 3)
| Объём жидкости, мл |
Концентрация раствора, г/дл |
Время истечения, с |
ηотн |
ηуд |
ηуд/С., дл/г |
| 8 |
0,7850 |
97,75 |
1,126 |
0,126 |
0.161 |
| 10 |
0,6280 |
96,31 |
1,109 |
0,109 |
0,174 |
| 13 |
0,4831 |
93,90 |
1,081 |
0,081 |
0,168 |
| 17 |
0,3694 |
92,12 |
1,061 |
0,061 |
0,165 |
На основании данных табл.5.8 построена зависимость ηприв - С, приведенная на рис. 5.8.

Рисунок 5.8. Зависимость ηуд
/С-С раствора сополимера индена с малеиновым ангидридом (СПЛ 3) в ацетоне
Таблица 5.9 – Данные определения вязкости растворов в ТГФ сополимера индена с малеиновым ангидридом(СПЛ 4), полученного из смеси I (см. табл. 5.3)
| Растворитель |
Объём жидкости, мл |
Концентрация раствора, г/дл |
Среднее время истечения, с |
ηотн. |
ηуд. |
ηуд/С., дл/г |
| ТГФ |
8 |
0,8098 |
126,53 |
1,0993 |
0,0993 |
0,1226 |
| 10 |
0,6478 |
125,27 |
1,0884 |
0,0884 |
0,1231 |
| 13 |
0,4983 |
123.31 |
1,0713 |
0,0713 |
0,1290 |
| 17 |
0,3811 |
122,12 |
1,0610 |
0,0610 |
0,1443 |
| ДМСО |
6 |
0,7331 |
106,54 |
1,0679 |
0,0679 |
0,0926 |
| 8 |
0,5498 |
104,41 |
1,0465 |
0,0465 |
0.0846 |
| 11 |
0,3999 |
103,41 |
1,0365 |
0,0365 |
0,0913 |
| 15 |
0.2933 |
100,98 |
1,0121 |
0.0121 |
0.0413 |

Рисунок 5.9 – Зависимость 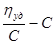 для растворов СПЛ 4 в ТГФ (1) и ДМСО (2). для растворов СПЛ 4 в ТГФ (1) и ДМСО (2).
Таблица 5.10 – Данные определения вязкости растворов в ДМСО сополимера, полученного из смеси II (см. табл. 5.3, СПЛ 5)
| Объём раствора, мл |
Концентрация раствора, г/дл |
Среднее время истечения, с |
ηотн. |
ηуд. |
ηуд/С., дл/г |
| 6 |
0,7331 |
106,54 |
1,0679 |
0,0679 |
0,0926 |
| 8 |
0,5498 |
104,41 |
1,0465 |
0,0465 |
0.0846 |
| 11 |
0,3999 |
103,41 |
1,0365 |
0,0365 |
0,0913 |
| 15 |
0.2933 |
100,98 |
1,0121 |
0.0121 |
0.0413 |

Рисунок 5.10 – Зависимость растворов в СПЛ 5
Далее были определены вязкости растворов, полученных в ампулах 1,3,4,6 (табл. 5.11 и 5.12) и 7,8 (табл. 5.13 и 5.14).
Таблица 5.11 – Данные определения вязкости растворов в ДМСО сополимеров индена с МА, полученного при соотношении инден: МА 90: 10 из смеси II в ампулах 1 и 3 (СПЛ 6 и 7)(смесь I` табл.5.3)
| № ампулы |
Объём раствора, мл |
Концентрация раствора, г/дл |
Среднее время истечения, с |
ηотн. |
ηуд. |
ηуд/С., дл/г |
| 1 |
6 |
0,8101 |
105,79 |
1,0603 |
0,0603 |
0,0744 |
| 8 |
0,6076 |
103,58 |
1,0382 |
0,0382 |
0,0629 |
| 11 |
0,4419 |
101,64 |
1,0187 |
0,0187 |
0,0423 |
| 15 |
0,3241 |
100,32 |
1,0055 |
0,0055 |
0,0170 |
| 3 |
6 |
0,8122 |
107,31 |
1,0756 |
0,0756 |
0,0924 |
| 8 |
0,6092 |
104,72 |
1,0496 |
0,0496 |
0,0806 |
| 11 |
0,4431 |
102,73 |
1,0297 |
0,0297 |
0,0658 |
| 15 |
0,3249 |
100,88 |
1,0111 |
0,0111 |
0,0327 |
На основании полученных данных была построена зависимость ηприв – С, приведенная на рис. 5.11.

Рисунок 5.11 – Зависимость ηприв – С растворов в ДМСО сополимера, полученного из смеси II (см. табл. 5.3).
Таблица 5.12 – Данные определения вязкости растворов в ДМСО сополимеров индена с МА, полученных из смеси III в ампулах 4 и 6 (СПЛ 8 и 9)
| № ампулы |
Объём жидкости, мл |
Концентрация раствора, г/дл |
Среднее время истечения, с |
ηотн. |
ηуд. |
ηуд/С., дл/г |
| 4 |
6 |
0,8101 |
105,15 |
1,0539 |
0,0539 |
0,0665 |
| 8 |
0,6076 |
103,43 |
1,0367 |
0,0367 |
0,0604 |
| 11 |
0,4419 |
101,71 |
1,0194 |
0,0194 |
0,0439 |
| 15 |
0,3241 |
100,52 |
1,0075 |
0,0075 |
0,0231 |
| 6 |
6 |
0,8153 |
107,94 |
1,0819 |
0.0819 |
0,1005 |
| 8 |
0,6115 |
105,61 |
1,0585 |
0,0585 |
0,0957 |
| 11 |
0,4447 |
103,14 |
1,0338 |
0,0338 |
0,0760 |
| 15 |
0,3261 |
101,56 |
1,0179 |
0,0179 |
0,0549 |
На основании полученных данных была построена зависимость ηприв – С, приведенная на рис. 5.12.

Рисунок 5.12 – Зависимость ηприв – С растворов в ДМСО сополимеров индена с МА, полученных из смеси III в ампулах 4 и 6
Таблица 5.13 – Данные определения вязкости растворов в ДМСО сополимера индена с МА, полученного из смеси IV из ампулы 7(СПЛ 10)
| Объём жидкости, мл |
Концентрация раствора, г/дл |
Среднее время истечения, с |
ηотн. |
ηуд. |
ηуд/С., дл/г |
| 1 |
2 |
3 |
4 |
5 |
6 |
| 6 |
0,1281 |
105,57 |
1,0581 |
0,0581 |
0,0716 |
| 8 |
0.6084 |
103,68 |
1,0392 |
0,0392 |
0,0644 |
| 11 |
0.4425 |
101,98 |
1.0222 |
0,0222 |
0,0502 |
| 15 |
0,3245 |
100,60 |
1,0083 |
0,0083 |
0,0256 |
На основании полученных данных была построена зависимость ηприв – С, приведенная на рис. 5.13.

Рисунок 5.13 – Зависимость ηприв – С, растворов сополимера индена с МА, полученного из смеси IV из ампулы 7
Таблица 5.14 – Данные определения вязкости растворов в ДМСО сополимера индена с МА, полученного из смеси IV из ампулы 8 (СПЛ 11)
| Объём жидкости, мл |
Концентрация раствора, г/дл |
Среднее время истечения, с |
ηотн. |
ηуд. |
ηуд/С., дл/г |
| 6 |
0,8091 |
104,98 |
1,0522 |
0,0522 |
0,0645 |
| 8 |
0,6068 |
103,31 |
1,0355 |
0,0355 |
0,0585 |
| 11 |
0,4413 |
101,54 |
1,0177 |
0,0177 |
0,0401 |
| 15 |
0,3236 |
100,42 |
1,0065 |
0,0065 |
0,0201 |
На основании полученных данных была построена зависимость ηприв – С, приведенная на рис. 5.14.

Рис. 5.14 – Зависимость ηприв – С, растворов сополимера индена с МА, полученного из смеси IV из ампулы 8 (СПЛ 11)
6. Обсуждение результатов эксперимента
6.1 Результаты сополимеризации
В ходе курсовой работы была проведена полимеризация кумарон-инденовой фракции (КИФ) с МА, добавленного в количестве 10 мол. % от непредельных соединений КИФ и 0,02 моль/л ПБ при 353 К. После выдерживания смеси в течение 3 ч и высаждения в петролейный эфир, выделили твёрдый осадок и смолу в соотношении 48:51 мас.% . Общий выход полимерных продуктов составляет ~17 % от суммы полимеризующихся соединений или ~ 7 % от исходной массы КИФ. Без добавления МА в аналогичных условиях продукты полимеризации выделить не удалось. Проведение в тех же условиях полимеризации ИФ с 10 мол. % МА (в среде толуола как модели КИФ) привело к получению около 40 %. В обоих случаях сразу же после введения МА наблюдалось образование мелкодисперсного осадка. Учитывая положительный результат сополимеризации КИФ и ИФ с МА продолжили изучение этого процесса в гомогенной среде в растворе ДО при 60ºС. Состав полимеризующейся смеси приведен в табл. 5.1, а изменение её объёма в ходе процесса в табл. 5.2. Полимеризацию проводили как с радикальным инициатором – пероксидом бензоила (ПБ), так и в присутствии ПБ и КО – Ti(OBu)4 (полимеризационные смеси I и II). В этих опытах получено 18,1 и 16,3 мас. % продуктов соответственно, что соответствует коэффициентам контракции 0,141 и 0,158. Эти величины отличаются между собой на 10 %, что можно считать практически одинаковым значением в пределах ошибки определения.
Согласно полученным результатам полимеризация смеси I протекает со скоростью 5,7 % /ч до конверсии около 10 мас. %, после чего несколько замедляется, и на отрезке от 10 до 18 мас. % скорость составляет 3,2 %/ч (рис. 5.1). В присутствии КО Ti(OBu)4 процесс протекает подобным образом, но его скорость несколько ниже: 4, 4 и 2,3 %/ч на первом и втором участках кривой, а перегиб наблюдается при конверсии около 13 мас. %. Наличие перегиба может быть связано как с ингибирующим действием растворителя, так и с замедлением реакции за счёт уменьшения количества МА (3 – 5 мол. % в точке перегиба) при условии образования чередующегося сополимера. Без ПБ продукты полимеризации не образуются.
Итак, введение данного КО при температуре 333 К не приводит к увеличению скорости процесса. Чтобы выяснить, как изменяется скорость процесса далее, проводили его до 22 ч (рис. 5.3). Оказалось, что через 5 – 6 ч процесс резко замедляется и в течение периода от 7 до 22 ч идёт со скоростью 0,4 %/ч.
Прямолинейная зависимость конверсии от времени отсекает на оси ординат отрезок, равный 17,9 %. Эта величина соответствует максимальному выходу СПЛ индена с МА состава 1:1 для реагирующей смеси, содержащей 10 мольн. % МА. Этот факт позволяет предположить радикальный процесс протекания чередующейся сополимеризации индена с МА и не противоречит сделанному ранее предположению о замедлении процесса за счёт исчерпания МА. Процесс идёт медленно, со скоростью 0,4 %/ч, что соответствует присоединению индена или других непредельных соединений, содержащихся в ИФ к уже образовавшемуся сополимеру индена с МА.
При проведении гетерогенной полимеризации в растворе толуола скорость процесса на начальных стадиях составляет 1,1 %/ч, т. е. она в 4-5 раз ниже, чем в растворе ДО. Затем процесс также несколько замедляется, но по достижении 13 % конверсии его скорость заметно возрастает, так что за 22 ч достигается примерно одинаковый выход – 24,8 % в ДО.
Ход зависимости S – t для гетерогенной сополимеризации (рис. 5.4) видимо, связан с тем, что в начале сополимеризация проходит в объёме раствора, а затем продолжается на частицах осадка, на границе раздела фаз и в объёме, что приводит к резкому повышению скорости процесса. При проведении процесса в условиях ступенчатого подъёма температуры от 80 до 100ºС показало, что выдержка реакционной смеси того же состава в толуоле (табл 5.5, смесь IV) при 80ºС и 3 ч при 90ºС позволяет получить 24,6 % продукта и не возрастает в ходе дополнительной выдержки при 100ºС в течение 3 ч.
Таким образом, подъём температуры до 90 - 100ºС позволяет сократить время проведения процесса в 3 – 4 раза, но не способствует увеличению выхода. Однако сополимеризация КИФ с МА, проведенная в том же температурном режиме, проходит в основном как гомогенный процесс с образованием незначительного количества осадка в ампуле. Кроме того, из реакционной смеси на основе КИФ удалось выделить 26,0 % продукта за 6 ч при 80 - 90ºС и 32,2 % при дополнительном нагревании до 100ºС (Выход рассчитывали, исходя из данных завода о содержании ~40 мас. % полимеризующихся компонентов в КИФ). Возможно, увеличение выхода при дальнейшем нагреве для КИФ связано с наличием компонентов, способных к гомогенной сополимеризации, например, стирола.
6.2 Характеристика продуктов полимеризации индена с малеиновым ангидридом
Для полученных продуктов полимеризации ИФ в присутствии МА определяли их характеристическую вязкость. Известно [40], что сополимеры индена с МА растворяются в ТГФ. Мы проверили также растворимость полученных нами образцов в хлороформе, ацетоне и ДМСО.
Оказалось, что хлороформ лишь частично растворяет подобные полимеры (С=0,055 г/дл). Кроме того, величина ηприв. для растворов в хлороформе с разбавлением увеличивается (табл. 5.6, рис. 5.5). Подобные зависимости описаны [41], причём отклонения от линейности проявляется, начиная с некоторой «критической концентрации», которая обычно ниже 0,1 г/дл. Аномалии критической вязкости могут быть связаны [42] с формой и размером макромолекулярных клубков, например, для полиэлектролитов. Однако они могут быть также обусловлены адсорбцией полимера на стенках капилляра, а также неточностью определения времени истечения. В нашем случае возможно специфическое взаимодействие жёсткой цепи, включающей бициклические звенья индена и циклические звенья МА, и хлороформа. На это указывает очень высокая величина приведённой вязкости, которая для СПЛ 1 примерно в 20 раз больше его вязкости в ацетоне (табл. 5.6, рис. 5.5 и 5.6). При этом сопоставление данных вязкости СПЛ 2 в ацетоне и ТГФ показывает, что они близки по величине и эти растворители примерно одинаково взаимодействуют с данным полимером. Величина характеристической вязкости СПЛ 2 в ацетоне составила 0,155 и 0,178 дл/г. Авторы [40] считают возможным использовать для определения молекулярной массы сополимера индена с МА константы, найденные для сополимера стирола с МА для ТГФ и ацетона
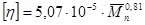 (6.1) (6.1)
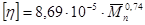 (6.2) (6.2)
По уравнению 6.1 и 6.2 молекулярная масса СПЛ 2 равна 24800 и 23800 в ацетоне и ТГФ. Таким образом, результаты определения молекулярной массы по данным вязкости в ТГФ и ацетоне хорошо совпадают и можно считать, что константы, приведенные авторами [43], хорошо подходят для определения молекулярной массы наших продуктов.
Для СПЛ 4 (табл. 5.9, рис. 5.9) получена характеристическая вязкость в ацетоне 0,154 дл/г и рассчитана молекулярная масса 24500. Как видно из этих результатов, молекулярная масса сополимеров, полученных при соотношении инден:МА 57:43 мол. % без и с Ti(OBu)4 практически одинакова.
Образцы сополимеров, полученные в растворе ДО, практически не растворяются в ацетоне и образуют мутный раствор в ТГФ, поэтому далее определение вязкости проводили для растворов в ДМСО. По СПЛ 5 измерена вязкость растворов в ТГФ и ДМСО (см. табл. 5.9 - 5.14). Найдено, что вязкость в ДМСО примерно в два раза ниже, чем в ТГФ. Для СПЛ 11 [η] в ТГФ составляет 0,145, а в ДМСО 0,08 дл/г. По уравнению (6.1) молекулярная масса этого образца равна 18600, поскольку ДМСО является более полярным растворителем и учитывая данные α для растворов полимеров в нём [43] приняли в нашем случае α=0,85. Тогда по величине молекулярной массы и α К=1,88· 10-5 дл/г. Эти величины констант далее испытали для расчёта молекулярной массы других образцов, вязкость которых измеряли в ДМСО. Данные определения характеристических вязкостей и молекулярных масс приведены в табл. 6. 1.
Таблица 6.1 – Данные определения характеристических вязкостей и молекулярных масс полученных сополимеров
| Образец |
Полимеризационная смесь |
Растворитель |
[η], дл/г |
Молекулярная масса |
| СПЛ 5 |
I (дил.) |
ТГФ |
0,145 |
- |
| ДМСО |
0,08 |
18600 |
| СПЛ 2 |
II (дил.) |
ДМСО |
0,084 |
19700 |
| СПЛ 6 |
I` (ампула 1) |
ДМСО |
0,0084 |
1310 |
| СПЛ 7 |
I`( ампула 3) |
ДМСО |
0,004 |
8230 |
| СПЛ 8 |
III (ампула 4) |
ДМСО |
0,002 |
3640 |
| СПЛ 10 |
IV (ампула 7) |
ДМСО |
0,04 |
8230 |
| СПЛ 11 |
IV (ампула 8) |
ДМСО |
0,035 |
7030 |
Образцы сополимеров, полученных в толуоле, имеют близкие значения характеристических вязкостей и молекулярных масс (табл. 5,9 – 5.14, 6.1, рис. 5,9 – 5.14), причём они в 2 -4 раза ниже, чем для сополимеров, полученных в ДМСО. Итак, проведение полимеризации в толуоле приводит к получению образцов с молекулярной массой, не превышающей 10000. Можно отметить тенденцию увеличения характеристической вязкости и молекулярной массы с ростом конверсии, например, для образцов (ампулы 4 и 7).
Выводы
1. Полимеризация индена с малеиновым ангидридом при температуре 60 – 100ºС протекает по радикальному механизму. Скорость полимеризации зависит от количества малеинового ангидрида, температуры и растворителей. Скорость реакции при 60ºС в растворе ДО при соотношении инден: МА 90:10 составляет 5,7 %/ч.
2. Сополимеризация в среде толуола протекает как гетерогенный процесс, который идёт с увеличением скорости.
3. Молекулярная масса сополимеров индена с МА, полученных в процессе сополимеризации в растворе ДО, составляет около 20000, а для гетерогенной сополимеризации в среде толуола получены образцы с молекулярной массой 2000 – 8000.
Список использованной литературы
1. Багдасарьян Х.С. Теория радикальной полимеризации. М.: Наука, 1966.–299с.
2. Говарикер. Полимеры. М. Мир, 1969 – 340стр.
3. Takebayashi M., Shingaki T., Ito Y. // Bull. Chem. Soc. Japan. 1953. V. 26. P. 475
4. Багдасарьян Х.С., Боровикова В.А. // Журн. физ. химии. 1961. Т. 35. С.2306.
5. Багдасарьян Х.С., Милютинская Р.И. Распад перекиси бензоила в различных растворителях // Журн. физ. химии. 1953. т. 27. Вып. 3. С. 420-432.
6. Nozaki K., Bartlet P. // J. Amer. Chem. Soc. 1946. V.68. № 22. P. 1686-?.
7. Bartlet P., Nozaki K. // J. Amer. Chem. Soc. 1946. V.68. № 22. P. 2299-?.
8. Cass W. // J. Amer. Chem. Soc. 1946. V.68. № 22. P. 1976-?.
9. Hartmann P., Sellers H., Turnball D. // J. Amer. Chem. Soc. 1947. V.69. № 22. P. 2416.
10. Кабанов В.А. Комплексно-радикальная полимеризация / В.А. Кабанов, В.П. Зубов, Ю.Д. Семчиков. – М.: Химия. –1987.– 256с.
11. Ikegami T., Hirai H. // J. Polym. Sci. 1970. V. 8. Pt. A-1. № 2. P. 463-482.
12. Голубев В.Б., Зубов В.П., Валуев Л.И. и др. // Высокомол. соед. 1969. Т. 11А. № 12. С. 2689-2694.
13. Tazuke S., Okamura S. // J. Polym. Sci. 1967. V. 5. Pt. A-1. № 5. P. 1083-1099.
14 Семчиков Ю.Д., Егорочкин А.Н., Рябов В.В. // Высокомол. соед. 1973. Т. 15Б. № 12. С. 893-895.
15. Ito T., Otsu T., Imoto M. // J. Polym. Sci. 1966. V. 4. Pt. B. № 1. P. 81-85.
Tazuke S., Okamura S. // J. Polym. Sci. 1967. V. 5. Pt. B. № 1. P. 95-99.
16. Соколов В.З. Инден-кумароновые смолы. М.: Металлургия, 1978.- 216 с.
17. Marechal E. // Bull. Soc. chim. France. 1969. № 5. P. 1459-1461.
18. Marechal E. // J. Macromol. Sci. 1973. A7. № 2. P. 433-460.
19. Metzner W., Wendish D. // Liebigs Ann. Chem. 1969. Bd. 730. S. 111-120.
20. Bass K.C., Hababsing P. // J. Chem. Soc. 1966. № 22. P. 2019-2023.
21. Коляндр Л.Я., Андреева В.С., Микитенко Л.Н. и др. // Кокс и химия. 1975. № 7. С. 28-30.
22. Neiser J. // Ropa a uhlie. 1959. № 6. S. 176-178.
23. Neiser J. // Ropa a uhlie. 1959. № 6. S. 176-178.
24. Sigwalt P. // C. r. Acad. Sci. 1961. № 24. P. 3800-3802.
25. Mizote A., Tanaka T., Higashimura T. et al. // J. Polym. Sci. 1966. V. 4, № 4. P. 869-879.
26. Eckhardt F., Heinze H.O. // Ztschr. analyt. Chemie. 1959. Bd. 730. № 1. S. 166-176.
27. Носков А.М. // Ж. прикл. спектроскопии. 1969. Т. 10 Вып. 4. С. 608-613.
28. Brause W. // Adhäsion. 1960. № 8. S. 393-396.
29. Андрианов Е.Г., Матвеева И.Е., Гавага В.С. // Кокс и химия. 1970. № 1. С. 47-49.
30. Е. В. Горохов. Ю. Б. Высоцкий, В. П. Королёв, С. И. Сохина. О. Н. Шевченко. Ю. В. Селютин. //Вiстник Донецького унiверситету, сер.А: Природничi науки. вип.1, 2003 – С.267-270.
31.. Русчев Д., Цачев А. // Химия и индустрия (НРБ). 1973. № 2. С. 83-88.
32 Литвинова Т.В. Пластификаторы резиновых смесей. М.: ЦНИИТЭ Нефтехим, 1971. - 87 с.
33. Ястржембская О.В., Андреева В.С. В кн.: Производство строительных изделий из пластмасс. Минск : Высшая школа, 1963. С. 159-164.
34. Sherwood P. // Chim. peintures. 1964. V. 27. № 10. P. 296-298.
35. Пат. (Австралия) № 283075. 1968.
36. Пат. (Австрия) № 232479. 1964.
37. Волков М.И., Ставицкий В.Д., Воронина В.Г. А.с. 210005 // Изобретения, промышл. обр., тов. знаки. 1968. № 5. С. 174.
38. Пат. (Яп.) № 7007. 1970.
39. Штерн Э., Тиммонс К. Электронная абсорбционная спектроскопия в органической химии. М.: Мир, 1974. - 295 с.
40. N., Ohno, S. Sugai//J.Macromol. sci., - Chem., 1990. Vol. A 27. P. 861 – 873.
41. Endo R., Hinokuma T., Takeda M. // J. Polim. Sci.1968. Vol. 6 A 2. №4. P. 675 – 685.
42. С. Р. Рафиков, С. А. Павлова, И.И. Твердохлебова. Методы определения молекулярных весов и полидисперсности высокомолекулярных соединений. Изд. АН СССР, М. – 1963.
43. Нестеров А.Е. Справочник по физической химии полимеров. Т. 1. К.: Наукова думка, 1984. 374 с.
Приложение
Была проведена статистическая обработка результатов измерений времён истечения растворителей и растворов полученных полимеров в данных растворителях. Результаты статистической обработки приведены ниже, в табл. 9.1, 9.2, 9.3, 9.4, 9.5 и 9.6.
Таблица 9.1 – Результаты расчета статистических характеристик определения времени истечения для хлороформа, ацетона, ТГФ и ДМСО
| № |
Хлороформ |
Ацетон |
| ti, с |
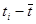 , с , с |
Ε |
ti, с |
, с |
ε |
| 1 |
76,36 |
0,12 |
0,10 |
86,82 |
-0,01 |
0,08 |
| 2 |
76,16 |
-0,08 |
86,83 |
0 |
| 3 |
76,28 |
0,04 |
86,92 |
0,09 |
| 4 |
76,19 |
-0,05 |
86,74 |
-0,09 |
| 5 |
76,22 |
-0,02 |
86,82 |
-0,01 |
| Σ |
381,21 |
- |
434,1 |
- |
|
76,21 |
- |
86,83 |
- |
| № |
ТГФ |
ДМСО |
| ti, с |
, с |
Ε |
ti, с |
, с |
ε |
| 1 |
115,04 |
-0,06 |
0,08 |
99,91 |
0,14 |
0,15 |
| 2 |
115,04 |
-0,06 |
99,85 |
0,08 |
| 3 |
115,11 |
0,01 |
99,75 |
-0,02 |
| 4 |
115,17 |
0,07 |
99,59 |
-0,18 |
| 5 |
115,15 |
0,05 |
99,76 |
-0,01 |
| Σ |
575,51 |
- |
498,86 |
- |
|
115,10 |
- |
99,82 |
- |
Из таблицы 9.1 видно, что для хлороформа время истечения 76,36 с для хлороформа не входит в доверительный интервал и его следует отбросить при расчёте , которое теперь составляет 76,21 с. Для ацетона все значения времени истечения входят в доверительный интервал, для дальнейшего расчёта вязкости принимаем =86,83 с. Для тетрагидрофурана все величины времён истечения входят в доверительный интервал и их следует учитывать при расчёте .
Таблица 9.2 – Результаты расчета статистических характеристик определения времени истечения раствора сополимера индена с малеиновым ангидридом, полученного при соотношении инден: МА 90:10 (без комплексообразователя) в хлороформе
| № |
С=0,056 г/дл |
С=0,043 г/дл |
| ti, с |
с |
ε, с |
ti, с |
с |
ε, с |
| 1 |
84,41 |
0,08 |
0,10 |
83,10 |
-0,16 |
0,19 |
| 2 |
84,52 |
0,03 |
83,24 |
-0,02 |
| 3 |
84,46 |
-0,03 |
83,30 |
0,04 |
| 4 |
84,55 |
0,06 |
83,38 |
0,12 |
| Σ |
337,94 |
- |
333,02 |
- |
|
84,49 |
- |
83,26 |
- |
| № |
С=0,032 г/дл |
С=0,024 г/дл |
| ti, с |
с |
ε, с |
ti, с |
с |
ε, с |
| 1 |
82,59 |
0 |
0,08 |
81,71 |
0,04 |
0,07 |
| 2 |
82,63 |
0,04 |
81,65 |
-0,02 |
| 3 |
82,51 |
-0,08 |
81,62 |
-0,05 |
| 4 |
82,61 |
0,02 |
81,71 |
0,04 |
| Σ |
247,73 |
- |
326,69 |
- |
|
82,59 |
- |
81,67 |
- |
В соответствии с величинами доверительных интервалов в таблице 9.2 все значения времени истечения раствора полимера входят в доверительный интервал и их следует учитывать при расчёте .
Таблица 9.3 – Результаты расчета статистических характеристик определения времени истечения раствора сополимера индена с малеиновым ангидридом, полученного при соотношении инден: МА 90:10 (без комплексообразователя) в ацетоне
| № |
С=0,087 г/дл |
С=0,068 г/дл |
| ti, с |
, с |
ε, с |
ti, с |
, с |
ε, с |
| 1 |
87,48 |
-0,10 |
0,07 |
87,29 |
-0,06 |
0,10 |
| 2 |
87,54 |
0 |
87,28 |
-0,07 |
| 3 |
87,56 |
0,02 |
87,38 |
0,03 |
| 4 |
87,59 |
0,05 |
87,42 |
0,07 |
| 5 |
87,54 |
0 |
87,36 |
0.01 |
| Σ |
437,71 |
- |
436,73 |
- |
|
87,54 |
- |
87,35 |
- |
| № |
С=0,051 г/дл |
С=0,038 г/дл |
| ti, с |
, с |
ε,с |
ti, с |
, с |
ε,с |
| 1 |
87,22 |
-0.12 |
0,10 |
87,23 |
-0,09 |
0,10 |
| 2 |
87,36 |
0,02 |
87,28 |
0,04 |
| 3 |
87,33 |
-.0,01 |
87,42 |
0,1 |
| 4 |
87,38 |
0,04 |
87,39 |
0,07 |
| 5 |
87,43 |
0,09 |
87,26 |
-0,06 |
| Σ |
436,72 |
- |
436,58 |
- |
|
87,34 |
- |
87,32 |
- |
Таблица 6.4 – Результаты расчета статистических характеристик определения времени истечения раствора сополимера индена с малеиновым ангидридом, полученного при соотношении инден: МА 57:43 (без комплексообразователя) в ацетоне
| № |
С=0,780 г/дл |
С=0,624 г/дл |
| ti, с |
, с |
ε, с |
ti, с |
, с |
ε, с |
| 1 |
96,09 |
0 |
0,13 |
95,36 |
0,08 |
0,12 |
| 2 |
95,91 |
-0,18 |
95,31 |
0,03 |
| 3 |
96,17 |
0,08 |
95,33 |
0.05 |
| 4 |
96,16 |
0,07 |
95,12 |
-0,16 |
| 5 |
96,13 |
0,04 |
95,29 |
0.01 |
| Σ |
480,46 |
- |
476,41 |
- |
| , с |
96,09 |
- |
95,28 |
- |
| № |
С=0,480 г/дл |
С=0,367 г/дл |
| ti, с |
, с |
ε,с |
ti, с |
, с |
ε,с |
| 1 |
93,23 |
-0,09 |
0,12 |
91,77 |
0,08 |
0,11 |
| 2 |
93,31 |
-0,01 |
91,95 |
0,10 |
| 3 |
93,32 |
0 |
91,79 |
-0,06 |
| 4 |
93,28 |
-0.04 |
91,78 |
-0.07 |
| 5 |
93,48 |
0,16 |
91,94 |
0,09 |
| Σ |
466,62 |
- |
459,23 |
- |
| , с |
93,32 |
- |
91,85 |
- |
Согласно данным таблицы 9.3, время истечения 87,48 с для концентрации 0,087 г/дл и 87,22 с для концентрации 0,051 г/дл не входят в доверительный интервал и их следует отбросить.
Анализ значений времён истечения и доверительных интервалов, приведенных в таблице 9.4 позволяет сделать заключение, что величины времён истечения 95,91 с при концентрации 0,7798 г/дл, 95,12 с при концентрации 0,6238 г/дл и 93,48 с при концентрации 0,4798 г/дл не входит в доверительный интервал и их следует отбросить.
Таблица 9.5 – Результаты расчета статистических характеристик определения времени истечения раствора сополимера индена с малеиновым ангидридом, полученного при соотношении инден: МА 57:43 (в присутствии бутоксититана) в ацетоне
| № |
С=0,785 г/дл |
С=0,628 г/дл |
| ti, с |
, с |
ε, с |
ti, с |
, с |
ε, с |
| 1 |
97,70 |
-0,05 |
0,10 |
96,35 |
0,04 |
0,13 |
| 2 |
97,83 |
0.08 |
96,22 |
-0,09 |
| 3 |
97,81 |
0,06 |
96,34 |
0,03 |
| 4 |
97,78 |
0,03 |
96,45 |
0,14 |
| 5 |
97,64 |
-0,11 |
96,20 |
-0,11 |
| Σ |
488,76 |
- |
481,56 |
- |
|
97,75 |
- |
96,31 |
- |
| № |
С=0,483 г/дл |
С=0,369 г/дл |
| ti, с |
, с |
ε,с |
ti, с |
, с |
ε,с |
| 1 |
93,94 |
0,04 |
0,09 |
92,27 |
0,15 |
0,11 |
| 2 |
93,85 |
-0,05 |
92,07 |
-0,05 |
| 3 |
93,97 |
0,07 |
92,05 |
-0,07 |
| 4 |
93,80 |
-0,10 |
92,08 |
-0,04 |
| 5 |
93,92 |
0,02 |
92,12 |
0 |
| Σ |
469,48 |
- |
460,59 |
- |
| , с |
93,90 |
- |
92,12 |
- |
По результатам, приведенным в таблице 9.5 можно сделать вывод о том, что времена истечения 97,64 с при концентрации 0,7850 г/дл, 95,45 с при концентрации 0,6280 г/дл, 93,80 с при концентрации 0,4831 г/дл и 92,27 с при концентрации 0,3694 г/дл не входят в доверительный интервал и их необходимо отбросить и не учитывать при расчёте .
Таблица 9.6 – Результаты расчета статистических характеристик определения времени истечения раствора сополимера индена с малеиновым ангидридом, полученного при соотношении инден: МА 57:43 (без комплексообразователя) в тетрагидрофуране
| № |
С=0,8006 г/дл |
С=0,6405 г/дл |
| ti, с |
, с |
ε |
ti, с |
, с |
ε, с |
| 1 |
132,55 |
-0,02 |
0,07 |
127,80 |
-0,07 |
0,09 |
| 2 |
132,46 |
-0,11 |
127,99 |
0,12 |
| 3 |
132,54 |
-0,03 |
127,88 |
0,01 |
| 4 |
132,67 |
0,10 |
127,81 |
-0,06 |
| 5 |
132,63 |
0,06 |
127,88 |
0,01 |
| Σ |
662,85 |
- |
639,36 |
- |
| ,с |
132,57 |
- |
127,87 |
- |
| № |
С=0,4927 г/дл |
С=0,3768 г/дл |
| ti, с |
, с |
Ε,с |
ti, с |
, с |
ε,с |
| 1 |
126,23 |
-0,06 |
0,08 |
123,29 |
0,08 |
0,07 |
| 2 |
126,34 |
0,05 |
123,17 |
-0,04 |
| 3 |
126,36 |
0,07 |
123,24 |
0,03 |
| 4 |
126,21 |
-0,08 |
123,12 |
-0,09 |
| 5 |
126,31 |
0,02 |
123,22 |
0,01 |
| Σ |
631,45 |
- |
616,04 |
- |
| ,с |
126,29 |
- |
123,21 |
- |
На основании таблицы 9.6 можно заключить, что в доверительный интервал не входят значения 132,46 с и 132,67 с для концентрации 0,8006 г/дл, 127,99 с для концентрации 0,6405 г/дл и 123,29 с и 123,12 с для концентрации С=0,3768 г/дл и их следует отбросить и не учитывать при расчёте .
Для полимеров, полученных методом дилатометрии, была также проведена статистическая обработка результатов определения времён истечения, данные которой приведены в таблице 9.7.
Таблица 6.7 – Результаты статистической обработки измерений времени истечения раствора в ТГФ СПЛ индена с МА, полученного методом дилатометрии при соотношении инден: МА 90:10 (без комплексообразователя)
| № опыта |
С=0,8098 г/дл |
С=0,7184 г/дл |
| t, с |
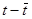 , с , с |
ε, с |
t, с |
, с |
ε, с |
| 1 |
126,48 |
-0,05 |
0,09 |
125,22 |
-0.05 |
0,06 |
| 2 |
126,48 |
-0,05 |
125,32 |
0,05 |
| 3 |
126,48 |
-0,05 |
125,32 |
0,05 |
| 4 |
126,63 |
0,1 |
125,27 |
0 |
| 5 |
126,60 |
0,07 |
125,20 |
-0,07 |
| Σ |
632,67 |
- |
626,33 |
- |
| ,с |
126,53 |
- |
125,27 |
- |
| № опыта |
С=0,5526 г/дл |
С=0,4226 г/дл |
| t, с |
, с |
ε, с |
t, с |
, с |
ε, с |
| 1 |
123,34 |
0,03 |
0,08 |
122,18 |
0.06 |
0,09 |
| 2 |
123,38 |
0,07 |
122,19 |
0,07 |
| 3 |
123,29 |
-0,02 |
122,06 |
-0,06 |
| 4 |
123,34 |
0,03 |
122,02 |
-0,10 |
| 5 |
123,20 |
-0,11 |
122,16 |
0,04 |
| Σ |
615,55 |
- |
610,61 |
- |
| ,с |
123,31 |
- |
122,12 |
- |
На основании данных таблицы 6.7 можно сделать заключение о том, что величины времён истечения 125,20 с для концентрации 0,7184 г/дл, 123,20 с для концентрации 0,5526 г/дл и 122,02 с для концентрации 0,4226 г/дл не входят в доверительный интервал и их необходимо отбросить и не учитывать при расчёте .
|
